Clinical and Pathological Findings of Korean Patients with Selenoprotein N-Related Myopathy
Article information

Abstract
Objective
This study investigated the clinical, pathological, and genetic characteristics of 5 Korean patients with selenoprotein N-related myopathy (SELENON-RM).
Methods
Five unrelated patients were genetically diagnosed with SELENON-RM by whole-exome or targeted gene panel sequencing. We then analyzed their clinical, pathological, and genetic spectra.
Results
The median age at symptom onset was 3 years (interquartile range, 2–10 years). The most common clinical finding was proximal muscle weakness in all 5 patients, followed by spinal scoliosis and respiratory distress in 4 patients and delayed motor development in 2 patients. Other uncommon clinical findings were winged scapula in one patient and cardiomegaly in one patient. Magnetic resonance imaging of muscles revealed that fatty replacement was predominant in the paraspinal muscles, adductors, semimembranosus, semitendinosus, long head of the biceps femoris, and medial gastrocnemius. Muscle biopsies in 2 patients showed type 1 predominance and multiple eccentric cores within the fibers. We identified 5 pathogenic variants of SELENON. The most common variant was the c.1574T > G variant in 5 alleles (50%) in 4 patients (80%).
Conclusion
In the first report of SELENON-RM in Korea, we identified 5 SELENON-RM patients and expanded existing knowledge on the clinical and genetic spectrum of these patients.
Introduction
Congenital myopathies are a clinically and pathologically heterogeneous group of genetic myopathies characterized by hypotonia, muscle weakness from birth, and a slowly progressive clinical course [1]. Congenital myopathies have been classically categorized based on muscle pathologies such as nemaline rods, cores, central nuclei, and selective hypotrophy of type 1 fibers [2,3]. The application of next-generation sequencing has made it possible to identify the genetic causes of congenital myopathies quickly and easily, and nearly 50 causative genes have been identified to date [4]. Recent advances in molecular diagnosis have shown that the relationship between genotype and phenotype is highly complex. The same gene can cause different phenotypes and muscle pathologies, and multiple genes can result in the same clinicopathological findings [5].
Selenoprotein N-related myopathy (SELENON-RM), caused by pathogenic variants of SELENON, is an autosomal recessive congenital myopathy. SELENON-RM clinically presents with slowly progressive proximal muscle weakness, spinal scoliosis, and respiratory insufficiency [6,7]. Multi-minicores are the main histopathological features of SELENON-RM, but congenital fiber-type disproportions and Mallory body-like inclusions have also been reported [7,8]. There have been large international case series in Europe [7], and some smaller case series in Asia [9–11]. However, there are no reports demonstrating the detailed clinical phenotypes of SELENON-RM patients in Korea. We evaluated the characteristics of SELENON-RM in a Korean population by analyzing clinical, pathological, and genetic data obtained from 5 unrelated patients with pathogenic variants in SELENON.
Materials and Methods
We reviewed the medical records from the myopathy database from January 2002 to December 2021 at Gangnam Severance Hospital. Five unrelated patients who had been diagnosed with SELENON pathogenic variants (MF27, MF88, MF1467, MF1481, and MF1949) were identified and included in this study. We previously reported one variant (c.619delC) as a novel pathogenic variant in SELENON in the MF88 patient, but did not present the detailed clinical and pathological phenotypes [12]. Clinical, laboratory, and pathology data were retrospectively obtained by reviewing patients' medical records. Data from before patients visited our institution were extracted from medical records submitted by patients from other hospitals. Clinical information included the age at symptom onset, motor development, muscle impairment, respiratory distress, scoliosis, and joint contractures. The laboratory analysis included serum creatine kinase (CK) levels. Whole-body muscle magnetic resonance imaging (MRI) was available for one patient (MF1467). Muscle biopsies were performed on 2 patients (MF27 and MF1467) at Gangnam Severance Hospital. Frozen muscle sections obtained from specimens were stained with hematoxylin and eosin, modified Gomori trichrome, and reduced nicotinamide adenine dinucleotide-tetrazolium reductase. The muscle biopsies of 2 other patients (MF88 and MF1481) had been conducted previously at other hospitals, and we analyzed their pathology reports.
This study was approved by the institutional Review Board of Gangnam Severance Hospital, Korea (IRB no. 3-2022-0011). All participants had previously provided informed consent for their involvement in this study and genetic analysis.
Results
The clinical and pathological phenotypes of the 5 patients with SELENON pathogenic variants are summarized in Table 1. All patients had 2 pathogenic variants of SELENON. All variants were classified as likely pathogenic or pathogenic according to the guidelines of the American College of Medical Genetics and Genomics/Association for Molecular Pathology (Table 2) [7,10,12–14].

Clinical Characteristics of Patients with Selenoprotein N-Related Myopathy

Pathogenic and Likely Pathogenic Variants in SELENON in the Present Study
A 5-year-old boy (MF27, II-1 in Fig. 1A) presented to our clinic with a 1-year history of respiratory insufficiency and proximal muscle weakness. He presented with delayed motor development; unsupported sitting was achieved at 12 months and independent gait at 18 months. When he was 4 years old, he was admitted to another clinic due to acute respiratory failure, and he was referred to our clinic 1 year later. He underwent a neurological examination that showed muscle hypotrophy and severe scoliosis with spinal rigidity. Laboratory studies revealed a CK level of 45 IU/L (reference value, < 135 IU/L). Needle electromyography was consistent with myopathic changes. A muscle biopsy was performed from the right vastus lateralis when the patient was 5 years old. It showed mild to moderate muscle size variation with type 1 fiber predominance, consistent with congenital fiber-type disproportions. One year later, he underwent tracheostomy and full-time non-invasive positive-pressure ventilation. At 15 years of age, the patient underwent corrective spinal surgery. He became unable to walk independently when he was 19 years old and was in a bedridden state. Whole-exome sequencing identified a compound heterozygous variant (c.13_22dup + c.1574T > G) in SELENON.
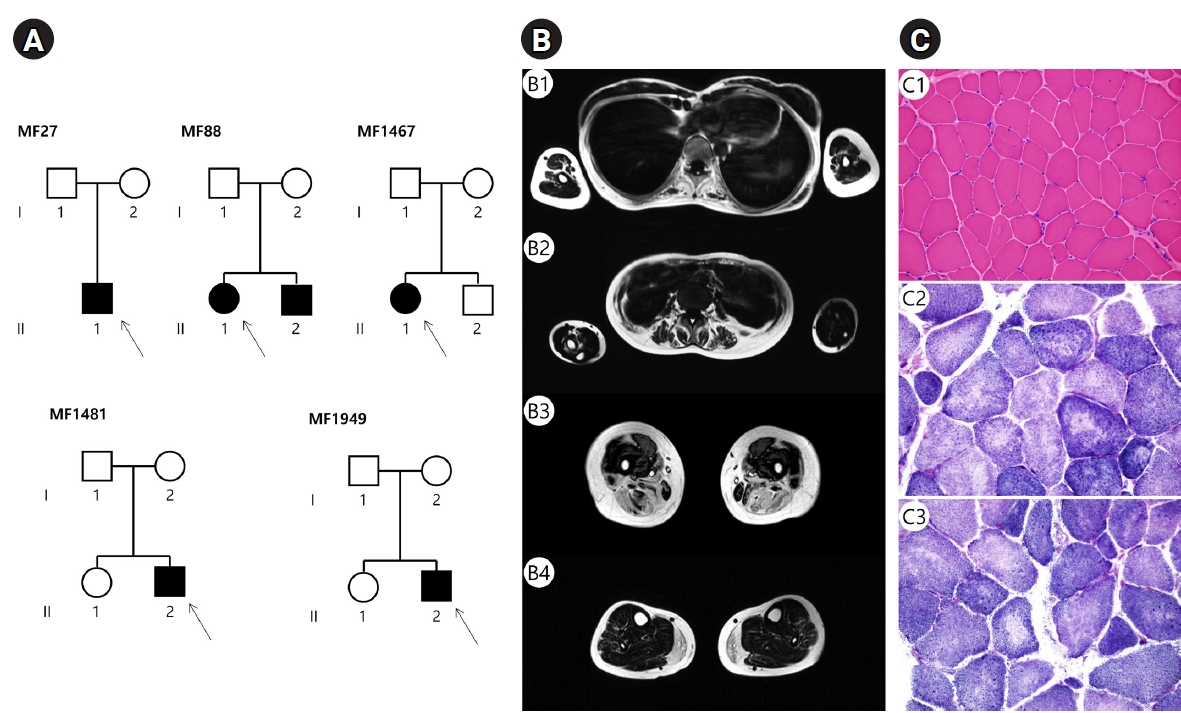
Pedigrees, muscle magnetic resonance imaging (MRI), and muscle biopsies in Korean families with SELENON-related myopathy. (A) Pedigrees of 5 families with SELENON pathogenic variants. Arrows indicate the probands (square, male; circle, female; filled, affected; unfilled, unaffected). (B) Muscle MRI findings of patient MF1467. Axial T1-weighted imaging revealed that fatty replacement was predominant in paraspinal muscles at the thoracic level (B1, B2), the adductor, semimembranosus, semitendinosus, and the long head of the biceps femoris at the thigh level (B3), and the medial gastrocnemius at the calf level (B4). (C) Muscle biopsy samples of the biceps brachii in MF1467. Mild muscle fiber size variation and little local necrotic fibers on hematoxylin and eosin staining (C1, ×200). Nicotinamide adenine dinucleotide staining, demonstrating multiple focal areas that are not stained (i.e., multiple eccentric cores) (C2, C3, ×400).
A 15-year-old girl (MF88, II-1 in Fig. 1A) was born to healthy parents at 40 weeks of gestation, weighing 2.5 kg. Her brother presented with pulmonary insufficiency and muscle weakness, and was previously diagnosed with muscular dystrophy. At 3 years of age, she experienced acute respiratory distress, but recovered within a few days. However, she had difficulty climbing stairs or getting up from sitting since the age of 5 years. Since the age of 11 years, she complained of spinal rigidity, scoliosis, and the aggravation of muscle weakness. The serum CK level was 66 IU/L, and needle electromyography showed generalized myopathy. According to the pathology report, she had undergone muscle biopsy when she was 14 years old from the left vastus lateralis, and it showed moderate-size variation in myofibers and a few degenerating fibers. When she was referred to our clinic at 15 years of age, she exhibited a high-arched soft palate, and a severe restrictive pattern on pulmonary function testing. She had forced vital capacity values below 35% of that predicted for height, which worsened every year. When she was 20 years old, she became unable to sit or walk alone. Targeted sequencing of 598 neuromuscular disorder genes identified a compound heterozygous variant (c.619del + c.1574T > G) of SELENON.
A 29-year-old woman (MF1467, II-1 in Fig. 1A) presented with a 10-year history of progressive muscle weakness. Her family history was unremarkable, and her initial psychomotor development was normal. She had an elongated face, bilateral facial paralysis, a high-arched palate, and winged scapula. A neurological examination revealed proximal and axial muscle weakness with the Gower’s sign. The serum CK level was mildly increased (182 IU/L). Needle electromyography revealed generalized myopathy. Whole-body MRI showed that fatty replacement and muscle atrophy were predominantly observed in the paraspinal muscles, adductors, semitendinosus, semimembranosus, the long head of the biceps femoris, and the medial gastrocnemius muscles (Fig. 1B). Histopathological examination of the left biceps brachii revealed increased variations in muscle fiber sizes and multiple eccentric cores within the fibers (Fig. 1C). At the age of 33 years, she could walk independently for about 100 m and did not complain of respiratory difficulty. Targeted sequencing of 598 neuromuscular disorder genes identified a compound heterozygous variant (c.234delC + c.565C > T) of SELENON.
A 19-year-old boy (MF1481, II-2 in Fig. 1A) was born to healthy parents at 40 weeks of gestation, weighing 3.2 kg. He presented with motor developmental delay, having achieved head control at 8 months and autonomous gait at 20 months. At the age of 13 years, he suffered from acute respiratory failure, and had been wearing a non-invasive ventilator. One year later, the patient underwent corrective spinal fusion surgery at another hospital. He underwent muscle biopsies from the quadriceps and paraspinal muscles at the ages of 13 and 14 years, respectively. Both muscle biopsy findings were recorded as nonspecific myopathic changes. When he visited our clinic at the age of 19 years, he was able to walk independently for up to 5 m. A neurological examination revealed diffuse muscle weakness and marked neck flexor weakness. Laboratory findings showed that the serum CK level had increased to 555 IU/L. Needle electromyography revealed generalized myopathy. Targeted sequencing of 598 neuromuscular disorder genes identified a compound heterozygous variant (c.619delC + c.1574T > G) of SELENON.
A 13-year-old boy (MF1949, II-2 in Fig. 1A) visited our clinic with pulmonary insufficiency and proximal muscle weakness. His family history was unremarkable. At the age of 10 years, he noticed difficulty climbing stairs and running. At the age of 11 years, he underwent general medical and orthopedic examinations. The patient exhibited severe contractures of the neck extensors with spinal scoliosis, and demonstrated weakness of the axial and proximal muscles. The serum CK level was 100 IU/L, and electromyography revealed generalized myopathy. According to his medical records, cardiomegaly was observed on chest radiography and pericardial effusion on echocardiography. The electrocardiogram demonstrated a normal sinus rhythm. Because of proximal weakness, including neck contracture with cardiac involvement, the patient was initially suspected of having Emery-Dreifuss muscular dystrophy. When he was 13 years old, he experienced acute respiratory distress requiring a tracheostomy with ventilator support. At the age of 21 years, he was able to walk up to 50 m with assistance and intermittent non-invasive ventilator use, especially at night. Targeted sequencing of 598 neuromuscular disorder genes identified a homozygous variant (c.1574T > G) of SELENON.
Discussion
We analyzed the genetic, clinical, and pathological spectra of 5 Korean patients with SELENON-RM. The present study identified 5 pathogenic variants in 5 unrelated Korean patients. Four variants were classified as pathogenic according to the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology and have been previously reported [7,12,15].
SELENON is an endoplasmic reticulum (ER) protein that plays an important role in oxidative stress protection and redox-related calcium homeostasis [8]. Although the precise pathogenic mechanism of SELENON-RM is not fully understood, SELENON deficiency affects excitation-contraction coupling in muscle [16], mitochondrial physiology, and energy metabolism [8]. In addition, it has been reported that depletion of SELENON increases susceptibility to metabolic disorders, including insulin resistance or lipotoxicity of saturated fatty acids, as a secondary change to ER stress [17]. Based on the pathophysiology of SELENON-RM, it is necessary to evaluate the changes in muscle mass and overall nutritional status as the disease progresses.
In agreement with previous studies [6,7,14,18], we found that in most patients, the onset of clinical symptoms was in the first decade, except for one patient who developed symptoms at the age of 19 years. According to the largest cohort studies with SELENON-RM [7], the first signs were noticed before the age of 15 years in all patients, and the proportion of patients with symptom onset after the age of 8 years was approximately 8%. In addition, all 5 patients predominantly had proximal muscle weakness. The most common phenotype in our study was spinal scoliosis with respiratory distress, consistent with the commonly known features of SELENON-RM. However, there are several differences between this study and previous reports. First, limb joint contractures or hyperlaxity, as one of the main clinical features, was not observed in our study. Second, the present study identified one patient who presented with a winged scapula without any spinal deformity (MF1467). There are no previous reports that the winged scapula is associated with SELENON-RM. Winged scapula may appear, but it is considered pseudo-winging due to dorsal hyperlordosis associated with spinal scoliosis [7]. Third, cardiac involvement is not known to be a clinical manifestation of SELENON-RM. If there is involvement, it is not as severe as mild ventricular hypertrophy, or it develops secondary to respiratory insufficiency [6,18]. However, we found a patient with a phenotype predominantly characterized by cardiac abnormality (MF1949).
Whole-body muscle MRI in the MF1467 patient showed that fatty replacement was predominant in the paraspinal muscles, adductor, semimembranosus, semitendinosus, long head of the biceps femoris, and medial gastrocnemius. These findings are consistent with the previously reported MRI data [14,19]. These selective muscle involvements may depend on the clinical distribution of muscle weakness and the presence of scoliosis.
Muscle pathology showed congenital fiber-type disproportion and multiple eccentric cores in MF27 and MF1467, respectively. These pathological findings are among the most common pathological findings of SELENON-RM [7,8]. However, we found medical records of nonspecific myopathic features of muscle biopsies in 2 patients (MF88 and MF1481) who underwent biopsies at other hospitals.
Our study identified 5 different pathogenic variants in SELENON. There were 3 frameshift variants (c.13_22dup, c.234del, and c.619delC), one nonsense variant (c.565C > T), and one missense variant (c.1574T > G). The most common variant was the c.1574T > G variant, which was found in 5 alleles (50%). This variant is located in exon 12, which modifies the nonpolar amino acid methionine into the acidic amino acid arginine. It was first reported in Japan [10]. Subsequently, a pathogenic variant was identified in a few cases in Asia [11,15]. However, in a large, wide-ranging case series in European countries, the c.1574T > G variant was found in only one of 132 patients. Ethnic variation in hotspots for SELENON or environmental factors may have contributed to these varied findings.
We did not find a correlation between the genotype and phenotype in patients with SELENON-RM. It is difficult to establish a rare disease, and we lack patients with the same pathogenic variants. A previous study reported that some exon 1 variants, including c.13_22dup, found in one of our patients (MF27), were associated with a severe phenotype [14]. Although this patient presented with clinical manifestations at the youngest age among our patients, it was not associated with disease severity. Null variants are usually correlated with a more severe phenotype than other types [14]. In our study, MF1467, with 2 null variants of SELENON (c.234del and c.565C > T), showed the mildest phenotype without respiratory distress and scoliosis. We identified the c.1574T > G missense mutation, which was the most common variant, in 5 alleles in 4 patients.
Conclusion
Our study represents the spectrum of clinical, pathological, and genetic findings in Korean SELENON-RM patients. We have outlined the clinical diagnostic clues of early axial and proximal muscle weakness, spinal scoliosis, and respiratory failure in SELENON-RM. In addition, we also observed one patient with a mild phenotype, suggesting that there is inter-individual variability in SELENON-RM. In conclusion, diagnosing and verifying the pathogenicity of genetic variants in SELENON-RM requires a comprehensive approach that integrates clinical characteristics, histological findings, muscle imaging, and genetic findings. This study is the first in Korea to analyze the various clinicopathological characteristics and genetic spectrum of SELENON-RM, which will contribute to improving the diagnosis and management of this condition and increasing disease awareness and recognition of its specific clinical characteristics.
Notes
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
The authors would like to thank the patients for their assistance with this work.