Introduction
Immunoglobulin G4-related disease (IgG4-RD) is an emerging condition that involves multiple organs, including the pancreas and biliary tract [1-3]. Neurological manifestations are relatively uncommon, but those that do occur can appear as isolated or combined neurological deficits, with pachymeningitis being the most frequently observed. IgG4-related pachymeningitis is characterized by hypertrophy; thus, neurological symptoms may arise not only from direct inflammatory infiltration, but also from the resulting mass effect [1,2,4]. In this report, we describe a rare case of IgG4-related hypertrophic pachymeningitis that presented with multiple lower cranial nerve palsies.
Case Report
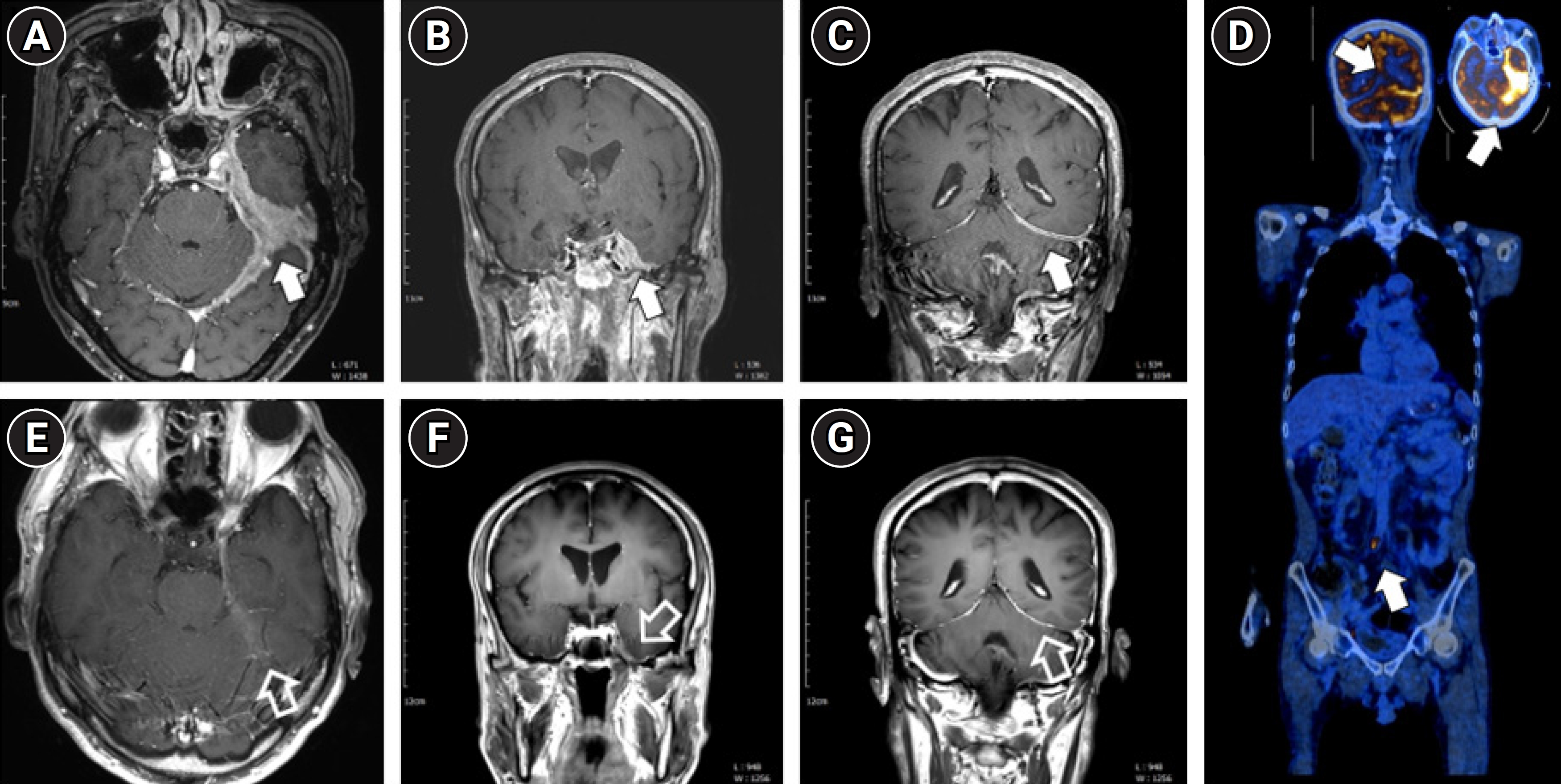
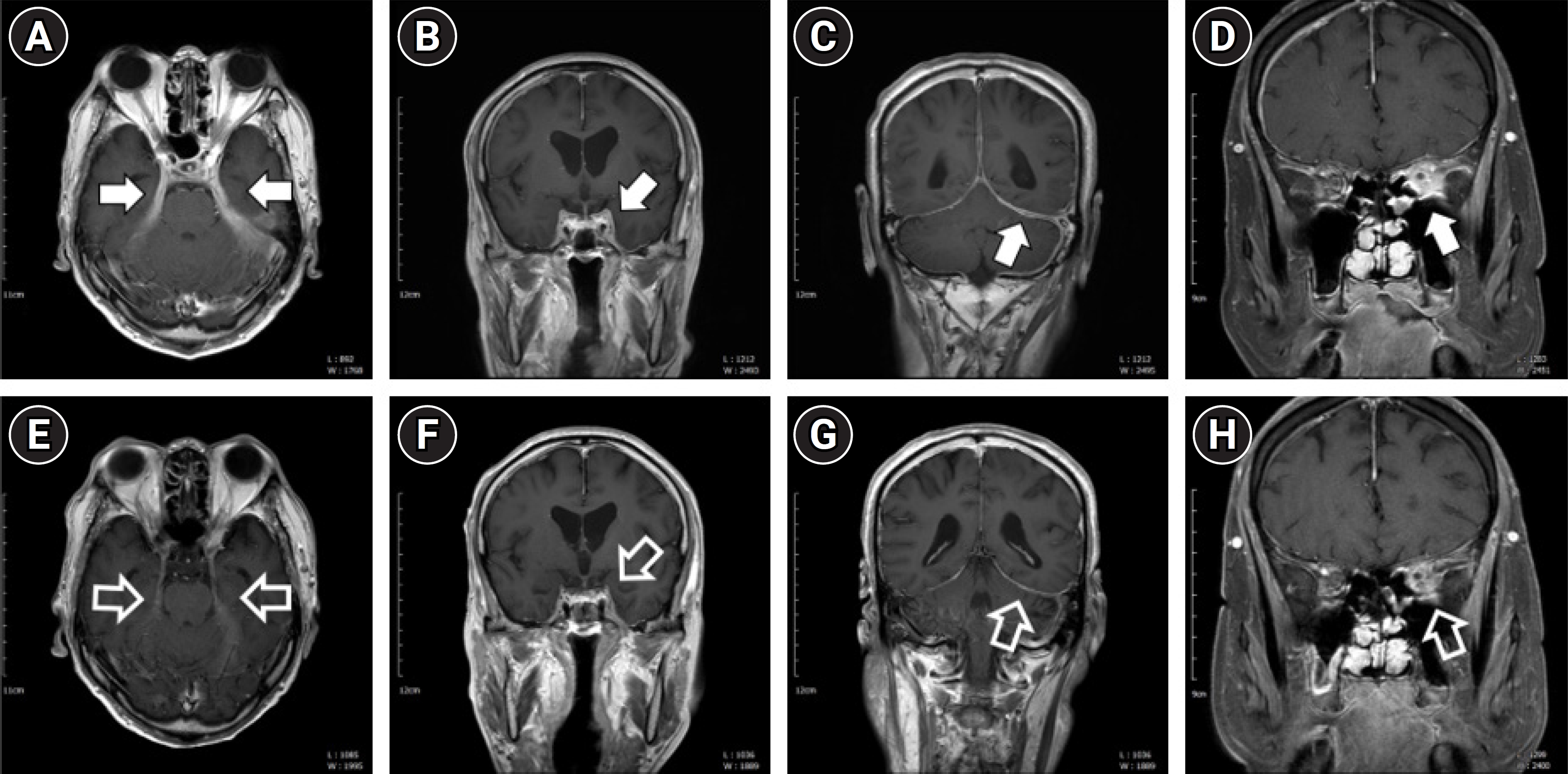
A 65-year-old man presented with headache, hoarseness, and dysphagia that had progressively worsened over the 5 months preceding hospital admission. He had first noticed a change in his voice and a slight tingling sensation while eating. These symptoms escalated until, after 2 months, he was unable to properly swallow solid food, resorting to a diet of mainly porridge. This was accompanied by symptoms of dry mouth and eyes. Upon neurological examination at admission, the patient exhibited a loss of gag reflex, palatal weakness, dysarthria with tongue deviation to the left, and weakness in the left sternocleidomastoid and trapezius muscles. These findings led to a diagnosis of multiple lower cranial nerve palsies, specifically affecting the left IX-XII cranial nerves. Other cranial nerve functions, limb motor and sensory function, and deep tendon reflexes were intact, with no pathologic reflexes. Consequently, we performed brain magnetic resonance imaging (MRI) to investigate the possibility of an organic brain lesion, such as a mass. MRI revealed diffuse dural thickening and enhancement along the left posterior fossa, tentorium, and temporal fossa, extending to the left internal auditory canal and Meckel cave, with suspected involvement of the left superior orbital fissure. Additionally, sinusitis was noted in the sphenoid, left maxillary, and ethmoid sinuses (Fig. 1). We conducted blood tests, cerebrospinal fluid (CSF) analysis, 18F-fluorodeoxyglucose positron emission tomography/computed tomography (FDG-PET/CT), and tissue biopsy, considering the differential diagnosis of IgG4-related disease as well as carcinomatous conditions and lymphoma. Laboratory results showed an elevated serum IgG level of 2,183 mg/dL (normal range, 700 to 1,600) and an increased serum IgG4 level of 218 mg/dL (normal range, 3 to 135). CSF analysis revealed a white blood cell count of 9/mm3, a protein level of 80.2 mg/dL, and a glucose level of 59 mg/dL. A tissue biopsy was performed on a polyp from the left ethmoid sinus. The biopsy results indicated chronic inflammation with moderate lymphoplasmacytic and some eosinophilic infiltration, without specific granuloma or vasculitis. Immunohistochemistry and special staining revealed up to 60 IgG4-positive cells per high-power field and an IgG4/IgG ratio of approximately 10% to 20%. FDG-PET/CT demonstrated abnormally high FDG uptake in the left posterior fossa, tentorium, and left temporal fossa, as well as mildly elevated FDG uptake in both pulmonary hila and interlobar lymph nodes. FDG uptake was also abnormally high in the proximal portion of the left renal artery and in the right common iliac artery (Fig. 1). Combining the test results with the patient’s clinical presentation, we diagnosed IgG4-related hypertrophic pachymeningitis with multi-system organ involvement, including the lacrimal and salivary glands, lungs, and retroperitoneum. The patient was treated with 1 g of intravenous methylprednisolone for 5 days, followed by 60 mg of oral prednisolone. Subsequently, the patient’s condition gradually improved; he was able to eat solid foods again, his voice returned to normal, and his symptoms of dry eyes and mouth also abated. He was then discharged from the hospital. Three months later, follow-up brain MRI showed improvement in the IgG4-related pachymeningitis (Fig. 1), and the patient’s serum IgG4 level had normalized to 50.3 mg/dL. However, the patient abruptly discontinued the oral steroid maintenance therapy and experienced recurrence 5 months after initial steroid therapy (Fig. 2). We attempted two cycles of intravenous steroid pulse therapy at 4-month intervals, along with oral maintenance therapy with 40 to 60 mg of steroids once a day and mycophenolate mofetil 500 mg twice a day. Despite these efforts, the treatment was not effective. Consequently, 16 months after initial symptom onset, we administered rituximab treatment (one cycle; 1 g of rituximab provided twice at a 2-week interval). Following this treatment, the patient’s symptoms and brain MRI findings displayed marked improvement, and all lower cranial nerve functions recovered (Fig. 2).
Discussion
IgG4-associated disease is an emerging clinical entity that can be categorized into two main subtypes. The first is IgG4-related autoimmune disease, which is marked by a predominance of autoantibodies of the IgG4 subclass. This group encompasses neuromuscular disorders such as myasthenia gravis with antibodies to muscle-specific tyrosine kinase and inflammatory neuropathies characterized by nodopathy/paranodopathy (involving contactin 1, contactin 1-associated protein 1, or neurofascin 155). Additionally, it includes conditions like pemphigus vulgaris, thrombotic thrombocytopenic purpura, autoimmune encephalitis (with antibodies against leucine‐rich glioma inactived‐1 [LGI-1], contactin-associated protein-like 2 [CASPR-2], or IgLON5), and membranous nephropathy. The IgG4 autoantibodies in this subtype are both target-specific and pathogenic, and serum IgG4 levels typically remain within the normal range. The second subtype is IgG4-RD. The role of IgG4 in this condition is still unclear, and fibrosis plays a key role in the disease pathology. Patients with IgG4-RD often exhibit elevated serum IgG4 levels with systemic involvement. IgG4-RD is also recognized as one of the causative diseases of hypertrophic pachymeningitis [5].
IgG4-RD can involve multiple organs in addition to the pancreas and biliary tract, and recognizing this multi-organ involvement is crucial for diagnosis. The 2019 classification criteria outlined by the American College of Rheumatology/European League Against Rheumatism categorize the disease into five domains: head and neck glands, chest, pancreas and biliary tree, kidneys, and retroperitoneum. These criteria, which we utilized for diagnosis, consider the involvement of various organs, serum IgG4 levels, histopathological findings, and immunostaining results [6,7]. Each organ’s involvement is verified through clinical examination or imaging techniques such as CT or PET scans, with a scoring system applied to each domain. A definitive diagnosis can be made when the cumulative score reaches 20 or higher. In the present case, PET/CT was employed to assess multi-organ involvement. Abnormal FDG uptake was detected not only in the brain but also in the bilateral pulmonary hila and interlobar lymph nodes. Involvement of the proximal portion of the left renal artery, the right common iliac artery, the chest, and the retroperitoneum was also confirmed. Additionally, clinical evidence of lacrimal and major salivary gland involvement was established. Consequently, a definitive diagnosis of IgG4-RD was made, based on a total score of 30 points (IgG4 level, 4 points; lacrimal/salivary gland involvement, 14 points; chest involvement, 4 points; retroperitoneal involvement, 8 points).
Since assessing the involvement of multiple organs is vital in the diagnosis of IgG4-RD, whole-body PET/CT serves as a valuable diagnostic tool. However, it may be challenging to reliably detect IgG4-related pachymeningitis using FDG-PET, as normal brain tissue exhibits substantial FDG uptake. Consequently, for the evaluation of brain lesions, brain MRI is the recommended imaging modality [1].
Neurological involvement is rarely observed in IgG4-RD, and this condition typically responds well to steroid treatment [1,2]. In cases of steroid-resistant IgG4-RD, rituximab therapy has been shown to be effective [8,9]. The present case demonstrates that steroids are an appropriate first-line treatment for IgG4-RD, and it also highlights the efficacy of rituximab for cases that are resistant to steroid therapy. In patients presenting with symptoms indicative of multiple lower cranial nerve palsies, particularly when differentiating from mass-like lesions [10], IgG4-RD should be considered in the differential diagnosis. To avoid diagnostic delays, serum IgG4 and IgG levels should be measured, and an examination for IgG4-specific histopathological features should be conducted.