Etiopathogenesis of Fibromyalgia
Article information

Abstract
Fibromyalgia is characterized by chronic widespread pain, and it is often accompanied by various symptoms such as fatigue, sleep disturbance, mood changes, cognitive dysfunction, and several somatic symptoms. The etiopathogenesis of fibromyalgia remains poorly understood, but it is thought to be caused by complex interactions among genetic predisposition, environmental factors, and biological factors. Emerging evidence suggests that central sensitization, which is characterized by impairment in the processes of pain perception, transmission, and modulation, plays an important role in fibromyalgia. Although various treatments have been used for fibromyalgia, patients still suffer from uncontrolled symptoms. Fibromyalgia still has many challenges to be solved. In this review, we discuss existing evidence on the etiopathogenesis of fibromyalgia.
Introduction
Fibromyalgia (FM), as the name implies, it is a disease accompanied by musculoskeletal pain throughout the body. This pain is chronic and widespread, with multiple tender points. In addition to pain, the disease is characterized by stiffness, fatigue, sleep disturbance, mood disorder, cognitive dysfunction, and various somatic symptoms. FM is the third most common musculoskeletal disease after low back pain and osteoarthritis. It is more common in women than in men, and its prevalence has been reported to be 2% to 5%, with some variation among studies. FM may also appear concurrently with other diseases such as infection, rheumatic disease, and neurological/psychiatric diseases, among others [1-4].
Although numerous studies have been conducted regarding the etiopathogenesis of FM, there are still many gaps in our understanding. There is no evidence that a single factor causes FM; instead, it is thought that FM is induced by a combination of genetic predisposition, environmental factors (such as physical and psychological stressors), and biological factors (such as sleep disorders, immunological abnormalities, neuroendocrine dysfunctions, psychiatric comorbidities, and neuroinflammation, among others) (Fig. 1). Central sensitization, which refers to abnormal hypersensitivity to pain due to errors in pain recognition and processing in the brain, is considered to play an important role in FM (Fig. 2) [5-7].
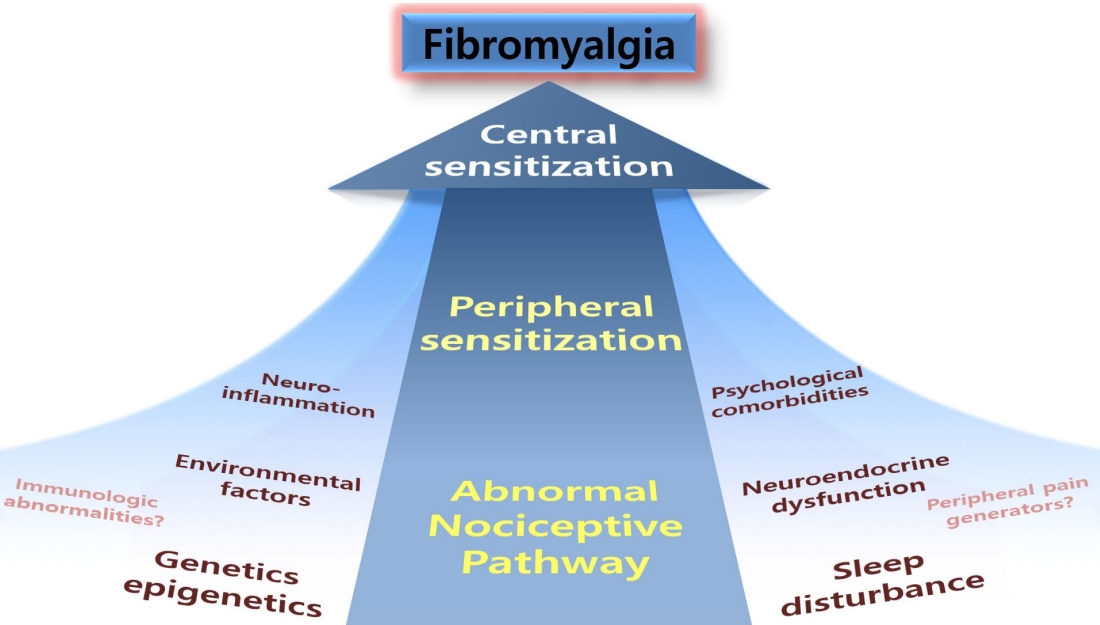
Etiopathogenesis of fibromyalgia.
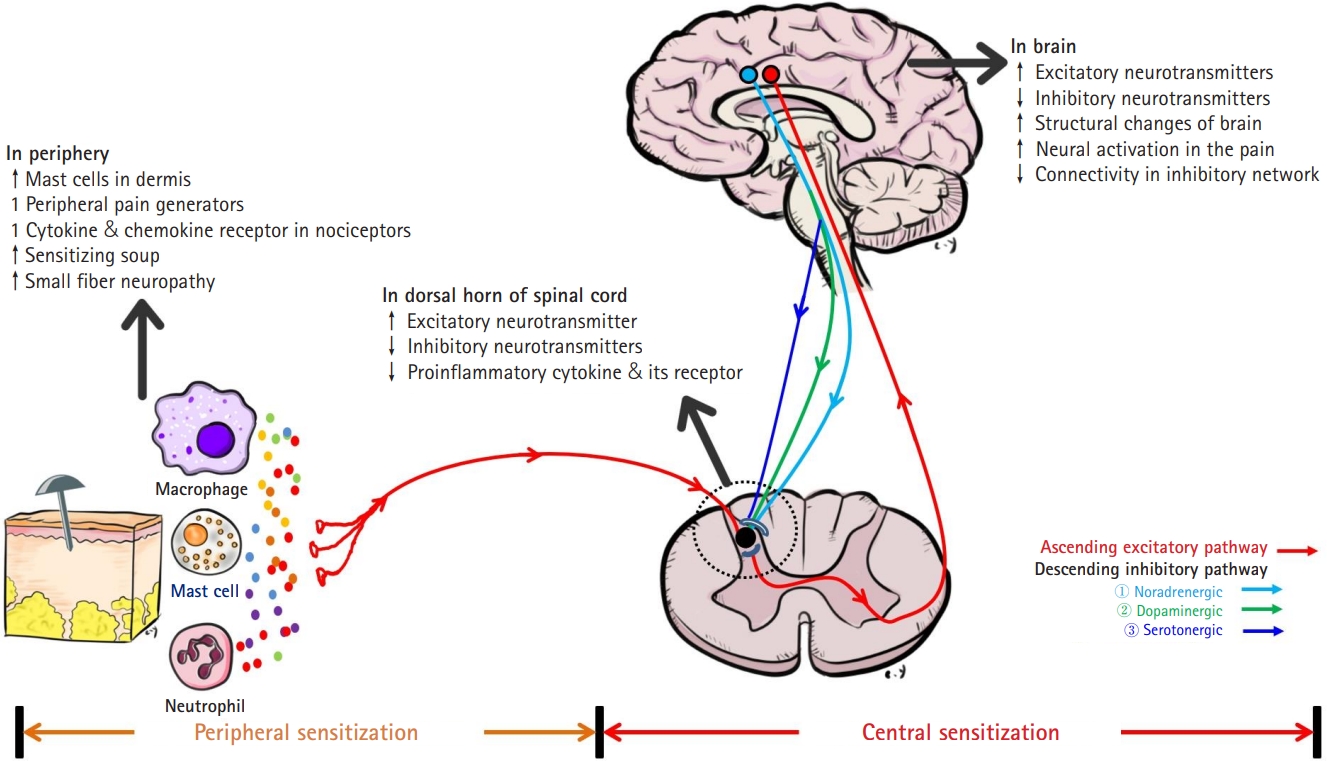
Pain mechanisms in fibromyalgia.
For the diagnosis of FM, the 2016 revised FM diagnostic criteria based on the 1990 American College of Rheumatology criteria are most commonly used [8]. However, since there is no specific biomarker for the diagnosis of FM and the diagnosis is made clinically, early diagnosis remains a challenge.
The diversity of clinical manifestations and underlying problems of each patient makes FM difficult to treat in clinical practice. In order to overcome these problems, clinicians have used combinations of non-pharmacological and pharmacological treatments, but the results remain disappointing. More effective and appropriate treatment is needed because many FM patients do not reach treatment targets and their daily lives are severely restricted. To this end, a better understanding of the etiopathogenesis of FM is required to facilitate the development of effective treatments.
Here, we aim to provide an overview of the etiopathogenesis of FM by reviewing studies to date.
Etiopathogenesis of Fibromyalgia
1) Genetics and epigenetics
(1) Genetic factors
The fact that epidemiological studies of FM show a distinct familial tendency suggests that genetic predisposition may play an important role in the pathogenesis of FM. The first-degree relatives of patients with FM were 8.5 times more likely to have FM than the first-degree relatives of patients with rheumatoid arthritis [9].
In a genome-wide association study with 116 families from the Fibromyalgia Family Study, the prevalence of FM in the general population was 2%, but its prevalence in siblings of patients with FM was 13.6%, indicating familial aggregation. Associations were observed in D17S2196 (empirical P [Pe]=0.00030) and D17S1294 (Pe=0.00035) in the chromosome 17p11.2-q11.2 region. This chromosome 17p11.2-q11.2 region matches the serotonin transporter (SLC6A4) and transient receptor potential vanilloid 2 (TRPV2) genes, which are potential candidate genes for FM [10]. In a genome-wide expression profiling study comparing 70 FM patients with 70 healthy controls, FM patients showed differential expression of 421 genes related to the pain pathway compared to the control group [11]. A genome-wide association study was conducted on 6,914 patients complaining of chronic widespread musculoskeletal pain registered with the UK Biobank and 242,929 control subjects. Loci for ring finger protein 123 (RNF123), which is involved in calcium regulation, and ATPase secretory pathway Ca2+ transporting 1 (ATP2C1) showed statistically significant associations in FM patients [12].
Genetic predisposition accounts for about 50% of disease susceptibility in chronic widespread pain, including FM [13]. Many genes associated with pain may also be involved in the pathogenesis of FM. Representative candidate genes include genes involved in the catecholaminergic, serotonergic, and gamma-aminobutyric acid (GABA)ergic pathways, μ-opioid receptors, and voltage-gated sodium channels [14-16].
In a single-nucleotide polymorphism (SNP) study, the GABA A receptor, beta 3 (GABRB3), trace amine associated receptor 1 (TAAR1), and guanylate binding protein 1 (GBP1) genes, which are associated with chronic pain, showed significantly different allelic frequencies in FM patients compared to the control group [14]. Next, we will examine the genes associated with neurotransmitters and receptors that play an important role in the pain pathway.
Serotonin and serotonin receptors play diverse roles in regulating the central nervous system. Dysregulation of the serotonergic system is involved in the pathogenesis of neurological disorders and pain, as well as psychiatric disorders such as anxiety, depression, schizophrenia, and autism [17-19]. Several studies have shown that specific genes related to the serotonergic system are also involved in the pathogenesis of FM. The frequency of carriers of the 102C allele of the 2A serotonin receptor gene (HTR2A) was significantly higher in patients with FM than in the control group, and FM patients had worse perceptions of environmental factors [20]. A meta-analysis also confirmed that the 102T/C polymorphism of the 5-HT2A receptor was linked to FM [21]. The frequency of the S/S genotype in the promoter region of the gene encoding the serotonin transporter (5-HTT) was significantly higher in patients with FM than in the control group, and depression and psychological distress were higher in the S/S subgroup [22].
Dopamine, epinephrine, and norepinephrine are catecholamine neurotransmitters that play an important role in various neurological disorders and pain control. Catechol-O-methyltransferase (COMT), a catecholamine-degrading enzyme, regulates the catabolism of catecholamine neurotransmitters. The activity of the COMT enzyme is regulated by major genotypic polymorphisms of alleles (Val/Val, Met/Met, Val/Met). Among homozygotes, the Val/Val genotype has the highest enzymatic activity and the Met/Met genotype has the lowest enzymatic activity, while heterozygotes, with the Val/Met genotype, exhibit moderate enzymatic activity [23,24]. The homozygous low activity (Met/Met) and heterozygous low activity (Val/Met) genotypes of COMT were predominant in FM patients compared to controls [25]. Among polymorphisms of the COMT gene, the Val158Met polymorphism has been studied the most, and has been found to be involved in various clinical symptoms including FM pain [26-28]. However, in a large-scale retrospective study examining the effect of the COMT gene on FM in 2,713 patients, no direct association between pain sensitivity-associated COMT haplotypes and FM pain was observed [29]. The COMT gene is the most studied gene in terms of FM susceptibility, but additional research is needed to clarify its association with FM.
Dopamine is involved in pleasure, motivation, and motor control, as well as pain modulation [30,31]. Dysregulation of the dopaminergic system is presumed to play an important role in the pathogenesis of FM [32]. FM patients showed a lower frequency of a dopamine D4 receptor polymorphism that had been reported to be related to novelty seeking than controls [33].
The dopamine receptor D3 (DRD3) Ser9Gly polymorphism was associated with lower thermal pain thresholds and diffuse noxious inhibitory control efficacy in patients with FM than in controls [34]. In a study evaluating the A118G rs1799971 polymorphism in the opioid receptor μ1 (OPRM1) gene, the 118G allele frequency was significantly lower in the FM patient group than in the control group, and pressure pain thresholds were also lower. This suggests that the 118G polymorphism may have a protective effect against FM [35]. In addition, it was found that the functional polymorphism of OPRM1 was associated with alterations in the fronto-parietal network, as well as with increased activation of the posterior cingulum during pain induction in patients with FM, suggesting that OPRM1 may play a role in pain processing [36].
Translocator protein (TSPO) is upregulated during glial activation in chronic pain patients. The high-affinity binding genotype (rs6971) of TSPO in FM patients was associated with higher pain intensity and more severe symptoms [37].
TRPV channels play an important role in pain signaling pathways. In a study of Korean FM patients, the GTA haplotype of TRPV2 played a protective role against FM. Polymorphisms of TRPV3 were reported to be associated with FM symptom severity [38]. In another study of Korean subjects, SNPs and haplotypes of the brain-derived neurotrophic factor (BDNF) gene, which is involved in the survival, growth, and differentiation of neurons during the development of the central and peripheral nervous system, have also been found to increase susceptibility to FM and contribute to symptoms [39].
In addition, in a large candidate gene association study, the TAAR1, regulator of G protein signaling 4 (RGS4), cannabinoid receptor 1 (CNR1), and glutamate ionotropic receptor ampa type subunit 4 (GRIA4) genes were associated with FM [14].
These studies show that genetic factors can influence both susceptibility to FM and the severity of symptoms. However, since genetic factors do not completely explain the pathogenesis of FM, additional research is needed not only on individual genes, but also on the effects of combinations of various genetic mutations and interactions with environmental factors.
(2) Epigenetics
Gene-environmental associations have recently attracted attention as an etiology of FM. Epigenetics is the study of how environmental factors affect gene operation without altering the DNA sequence. Unlike genetic changes, epigenetic changes are reversible and can alter the expression of DNA sequences through mechanisms such as DNA methylation, histone modification, and non-coding RNA. It has been reported that these epigenetic changes in pain-related regions frequently occur during inflammation and nerve damage [40-42].
In a study examining genome-wide methylation patterns of female FM patients, BDNF, N-acetyltransferase 15 (NAT15), histone deacetylase 4 (HDAC4), protein kinase C, alpha (PRKCA), reticulon 1 (RTN1), and protein kinase, cGMP-dependent, type I (PRKG1) were identified as genes associated with differentially methylated sites in patients [43]. In a study examining changes in DNA methylation profiles, FM patients showed 1,610 differentially methylated positions, which were related to DNA repair pathways, mitochondria-related processes, synaptic signaling, MAPK signaling pathway, regulation of actin cytoskeleton, and focal adhesion. It was related to the cytoskeleton and focal adhesion. FM patients also showed hypomethylation DNA patterns in regions rich in genes involved in stress response and DNA repair/free radical clearance [44]. Patients with FM and chronic fatigue syndrome had significantly higher serum BDNF levels and lower BDNF DNA methylation than the control group. This was related to patients’ symptoms and widespread hyperalgesia [45].
MicroRNAs are short non-coding RNA molecules composed of approximately 20 to 22 nucleotides and can regulate gene expression in disease processes and physiological pathways. About 30% of human genes are regulated by microRNAs, and each microRNA can repress hundreds of genes [46,47]. In a study evaluating the genome-wide profile of microRNAs in the cerebrospinal fluid (CSF) of patients with FM, the expression levels of nine microRNAs (miR-21-5p, miR-145-5p, miR-29a-3p, miR-99b-5p, miR-125b-5p, miR-23a-3p, 23b-3p, miR-195-5p, miR-223-3p) were significantly lower than those in the control group. Among them, miR-145-5p showed a statistically significant positive correlation with Fibromyalgia Impact Questionnaire pain and fatigue scores [48]. In a study examining 374 circulating microRNAs in 20 female FM patients and healthy women, those with FM had lower levels of seven microRNAs (miR-103a-3p, miR-107, let-7a-5p, miR-30b-5p, miR-151a- 5p, miR-142-3p, and miR-374b-5p) and higher levels of miR-320a. Among them, miR-103a-3p showed a correlation with pain and sleep quantity in patients with FM, and miR-320a and miR-374b-5p showed inverse correlations with pain and the pain threshold, respectively [49].
The pathogenesis of FM is presumed to be caused by the complex interaction of various factors. Epigenetics is a very attractive possible explanation for the interaction between genetic predisposition and environmental factors [50], and it seems to be an area that requires further research.
2) Environmental factors
The environmental factors presumed to be involved in the pathogenesis of FM can largely be divided into physical and psychological stressors. Physical stressors include infection, repeated stimulation, mechanical/physical trauma, surgery, and accidents, among others, and psychological stressors include chronic stress, mental abuse, emotional trauma, and sexual abuse. These environmental factors are presumed to contribute to the pathogenesis of chronic pain, such as FM, through stimulation of the neuro-endocrine system [51-55].
In animal studies using rats and mice, repeated exposure to cold stress, swim stress, and sound stress induced thermal hyperalgesia and persistent deep mechanical hyperalgesia, which were relieved by antidepressant treatment [56-59]. In addition, repeated forced swimming stress reduced basal and pain-evoked release of GABA in the spinal cord [60]. Another study confirmed that the use of gabapentin and pregabalin inhibited the secretion of glutamate, an excitatory neurotransmitter [61]. Blocking N-methyl D-aspartate (NMDA) receptors in the central nervous system has been shown to block exercise-induced hyperalgesia [62].
Some infections, such as hepatitis C virus, hepatitis B virus, human immunodeficiency virus, and Borrelia burgdorferi, have been reported to be associated with the development of FM [63]. A cohort study conducted on the prevalence of FM 10 years after a Giardia lamblia outbreak was recently published. The prevalence of FM was approximately three times higher in the group exposed to G. lamblia, and FM was associated with irritable bowel syndrome and chronic fatigue. Although this result alone cannot prove a causal relationship, there did seem to be a close relationship [64].
Physical trauma is a trigger for FM and chronic pain, but it is difficult to prove a causal relationship. In some studies, the prevalence of FM after a car accident was reported to be between 1% and 22% [65-67]. In addition, physical stressors such as surgery, terrorism, or war experiences contribute to the onset of FM [68-70]. Stress is presumed to play an important role to the extent that FM is considered a stress-related disease, but this may vary depending on intensity, type, and exposure time of stress. In a community-based study, significant tenderness in FM was strongly associated with psychological distress and somatization [71]. In the British Birth Cohort Study, which was conducted from 1958 onward, chronic widespread pain in adulthood was more common among participants whose childhood experiences included hospitalization due to a traffic accident, residence in institutional care, maternal death, and familial financial hardship [72]. In addition, sexual or emotional assault/abuse can contribute to the onset of FM [73,74].
These psychological/physical stressors can cause various changes in the body. In the normal response to stress, the autonomic nervous system (ANS) and the hypothalamic-pituitary-adrenal (HPA) axis are activated [75-78]. However, if stress is excessive or lasts for a long time, dysregulation occurs, leading to FM and other chronic diseases [79-81]. Chronic stress can lead to neuroendocrine abnormalities such as HPA axis dysregulation, which can lead to a relative decrease in cortisol. It has been reported that metyrapone-induced hypocortisolism significantly lowers pain thresholds and amplifies the temporal summation of pain [82]. It was confirmed that taking 40 mg of orally administered hydrocortisone significantly reduced capsaicin-induced pain and neurogenic hyperalgesia, such as that in response to pinprick stimuli [83]. In addition, it has been reported that negative emotions such as anger and sadness directly amplify pain regardless of the presence or absence of FM [84].
3) Biological factors
(1) Dysfunction of the neuroendocrine system
The HPA axis is an important component of adaptive responses to stress, and dysfunction of the HPA axis has been found in patients with FM [85]. The HPA axis regulates neurotransmitters through interactions with the brain's serotonergic, noradrenergic, and dopaminergic systems, as well as stress. The final product, cortisol, acts on various tissues, among which the hypothalamus is an important target and can regulate the expression of various neurotransmitters [86,87]. Plasma cortisol levels in patients with FM have shown various results depending on the study, but there are many reports of dysregulation of circadian changes. FM patients have a lower cortisol response to corticotropin-releasing hormone (CRH) than normal controls [75]. In a study using patients with rheumatoid arthritis as a control group, a significant loss of diurnal variation in plasma cortisol levels was observed in patients with FM [88]. Another study reported that patients with FM had less diurnal fluctuation in cortisol than normal controls, indicating that the elasticity of the HPA axis had been lost [89]. In patients with FM, the concentration of CRH in the CSF was associated with pain [90]. Chronic stress also reduced total cortisol release by decreasing adrenal reactivity in patients with FM [91].
Growth hormone is mainly secreted during non-rapid eye movement (NREM) sleep, and is involved in not only growth, but also repair of micro-damage to muscle tissue. Growth hormone also promotes the production of Insulin-like growth factor 1 in the liver, and it has been found that both of these hormones are present in lower levels in patients with FM than in controls [92,93]. However, it is not yet clear whether these changes affect the development of FM or whether they are secondary changes caused by sleep disturbance in FM.
The ANS maintains homeostasis largely by regulating balance between the sympathetic nervous system and the parasympathetic nervous system. In FM, abnormal findings in the sympathetic nervous system are observed, contributing to several clinical features. Sympathetic hyperactivity is accompanied by increased emotional distress and decreased heart rate variability in FM. FM patients showed decreased nighttime heart rate variability, which indicated sympathetic predominance, a feature significantly correlated with FM symptoms such as pain intensity, constipation, and depression [94]. Overactivation of the sympathetic nervous system is associated with several symptoms of FM, but it remains unclear whether it is the cause.
(2) Sleep disturbance
Patients with FM often complain of feeling tired and not refreshed when they wake up in the morning, even after sleeping for a long time. Daytime sleepiness is also frequent. Sleep disturbance is very common in FM, occurring in about 90% of patients [95], and it may simply be a symptom of FM, but it can also be viewed as an etiological factor. There is evidence that sleep disturbance usually precedes the onset of pain [96].
An 11-year prospective cohort study targeting the general population reported that poor sleep, obesity, and chronic disease could predict widespread chronic pain [97]. In early studies, patients with FM exhibited NREM sleep disturbance, but even in healthy subjects, sleep disturbances have been found to induce muscle pain and tenderness similar to FM [98,99]. In a longitudinal study of 12,350 Norwegian women who had never been diagnosed with FM, 327 were diagnosed with FM on follow-up. There was a dose-dependent association between sleep problems and the risk of FM, and the adjusted relative risk of FM was reported to be 3.43 (95% confidence interval, 2.26 to 5.19) in women with a high frequency of sleep disturbance [100]. These findings strongly suggest that sleep disturbance may contribute to the onset of FM.
Sleep disturbance is associated with the exacerbation of various FM symptoms, including pain severity, tenderness point, fatigue, and depression [101-106]. Sleep also plays an important role in the pain pathway. Even in healthy individuals, sleep disorders inhibit the descending inhibitory pain pathway [107-109].
The mechanism by which sleep disturbance contributes to the exacerbation of symptoms of FM, especially pain, is as follows. First, In patients with FM, it is often observed that the alpha activity abnormally intrudes into the delta activity region in slow-wave sleep (stage 3 of NREM sleep). Slow-wave sleep has been found to reduce pain through synaptic downscaling [110,111]. Disrupted slow-wave sleep not only increases the pain response by lowering the musculoskeletal pain threshold, but is also associated with FM-like symptoms such as discomfort, fatigue, and decreased energy [101]. Therefore, it is presumed that alpha activity interferes with the pain suppression effect of normal delta activity, resulting in increased pain in FM [112]. Second, the thalamus plays an important role in sensory transmission pathways. Meanwhile, the thalamocortical circuit controls sensory information and NREM sleep [113]. In FM patients, reduced connectivity has been observed in the thalamus and premotor areas, right insula and primary sensorimotor areas, and supramarginal and prefrontal areas [114]. This reduced connectivity of the thalamic pathway or dysfunctional primitive thalamus lowers the restorative function of sleep, which can trigger somatic and psychological symptoms of FM [115]. Third, substance P (SP) was found to cause sleep disturbance in animal studies. Elevation of SP in plasma and CSF was observed in FM patients, suggesting that it may be a possible cause of sleep disturbance in FM patients [116-118]. Fourth, sleep disturbance can promote nociception through an increase in proinflammatory cytokines, such as interleukin 6 (IL-6) [119]. Last, sleep disturbance is also closely related to other fibromyalgia symptoms. The pain caused by fibromyalgia activates the sympathetic nervous system and causes various symptoms, which also reduces sleep efficiency, and eventually has adverse effects each other [120].
Both experimental and epidemiological studies suggest that sleep dysfunction can cause FM and is also closely associated with various FM symptoms. Conversely, FM can further promote sleep disorders, creating a vicious cycle through mutual influence.
(3) Psychiatric and psychological comorbidities
In total, 30% to 60% of FM patients have accompanying psychiatric disorders, such as anxiety or depression [121,122]. In a recent meta-analysis, the most common comorbidities in patients with FM were depression and major depressive disorder, with prevalence rates of 43% and 32%, respectively [123]. On functional magnetic resonance imaging (fMRI) in patients with FM, depression was associated with the magnitude of pain-evoked neuronal activation in brain regions associated with affective pain processing (the amygdala and contralateral anterior insula) [124]. Depression and FM are highly correlated in terms of genetic predisposition. A serotonin transporter promoter region (5-HTTLPR) polymorphism associated with anxiety-related personality traits has also been observed in FM [125]. Forebrain regions related to pain control include the limbic system, which is also involved in stress response and mood regulation [126]. In addition, HPA axis abnormalities, microglial activation, and proinflammatory cytokines, which are involved in the pathogenesis of FM, may be commonly involved in the pathogenesis of depression [127-131]. Various symptoms of FM severely interfere with the patients’ daily and work life, which can lead to additional psychosocial problems, such as anxiety and social isolation.
(4) Immunological abnormalities
Recent studies have reported that immunological mechanisms may be involved in the pathogenesis of FM. When serum immunoglobulin G (IgG) extracted from FM patients was intraperitoneally injected into mice, mechanical and cold hypersensitivity, reduced locomotion, reduced skin innervation, and increased nociceptor excitability were observed in the mice. In addition, biopsies showed that the patients’ IgG was bound to satellite glial cells in the mouse lumbar dorsal root ganglia (DRG), and showed higher immunoreactivity than IgG from healthy individuals. The activation of satellite glial cells induces chronic pain through increased neural activity. This suggests that autoantibodies from patients with FM can bind to antigens in the DRG of mice without systemic inflammation [132].
It has also been reported that antibodies to 5-hydroxytryptamine (5-HT), gangliosides, and phospholipids are detected at high frequency in patients with FM and chronic fatigue syndrome [133,134]. FM was confirmed to be associated with thyroid disease. Thyroid disease can exacerbate the symptoms of FM, and the frequency of thyroid autoantibodies has been confirmed to be high in FM patients [135,136]. Some studies have reported that immunological mechanisms may play an important role in FM, but overall evidence is lacking.
(5) Neuro-inflammation
A) Cytokines
Several studies have reported that proinflammatory cytokines are involved in the pathogenesis of FM. Serum concentrations of proinflammatory cytokines, IL-6, IL-8, IL-1β, and tumor necrosis factor α (TNFα) are increased in FM, while the concentrations of anti-inflammatory cytokines are decreased [137-140]. In a recent meta-analysis, the levels of TNF-α, IL-6, and IL-8 (proinflammatory cytokines) and IL-10 (an anti-inflammatory cytokine) were significantly higher in peripheral blood in FM patients than in healthy controls. This study suggests that cytokines may be involved in the pathogenesis of FM [141]. Peripheral nociceptors express various cytokine and chemokine receptors [142]. When these are stimulated, neuronal activity is activated and can play an important role in peripheral sensitization [143,144]. Spinal dorsal horn neurons also express various proinflammatory cytokine receptors [145]. Proinflammatory cytokines can enhance excitatory synaptic transmission in neurons of the spinal cord and suppress inhibitory synaptic transmission [141]. Patients with FM exhibit higher IL-8 levels in CSF than healthy controls [146]. IL‐8 is synthesized in microglial cells and astrocytes and is involved in pain control [147]. Blocking IL-8 receptors relieved pain in a mouse model [148]. This suggests that IL-8 may also be an important target for treatment in FM patients.
Proinflammatory cytokines can induce the sensitization of peripheral neurons by increasing reactivity to nitric oxide and prostaglandin E2 [149]. It may also be involved in the pathogenesis of FM by affecting the HPA axis, the sympathetic nervous system, and immune cells [150,151]. Although several studies on cytokines were limited by various types of bias, they generally suggested that cytokines can contribute some extent to the pathogenesis of FM.
B) Neuropeptides
Neuropeptides are small chains of amino acids synthesized and secreted by neurons, and are defined as various neuroactive substances responsible for communication between neighboring neurons. More than 100 different neuropeptides have been identified that regulate nerve activity and function in tissues such as the intestine, muscle, and heart [152].
Nerve growth factor (NGF), calcitonin gene related peptide (CGRP), SP, glutamate, serotonin, norepinephrine, GABA, and dopamine are representative neuropeptides related to pain. They play a very important role in peripheral sensitization and central sensitization, which will be described later. Adrenomedullin, neurokinins, and vasoactive intestinal peptide are also neuropeptides contributing to pain pathways [153].
SP is secreted from the terminals of specific sensory nerves and contributes to the inflammatory response and pain in the brain and spinal cord, as well as peripheral nerves. When the SP level increases, the sensitivity to pain increases as well. It was found that the concentration of SP in the CSF of patients with FM was two to three times higher than that of normal controls [154,155].
CGRP regulates motor, sensory, and integration systems in the central nervous system, as well as synaptic transmission through inhibition of acetylcholine esterase expression at the neuromuscular junction in the peripheral nervous system. It is a neuropeptide that plays a very important role in peripheral and central sensitization in the pain pathway [156-158]. Serum CGRP and CGRP receptor protein levels were significantly elevated in FM patients compared to healthy controls [159].
Glutamate is a representative excitatory neurotransmitter that plays an important role in brain metabolism [160]. Glutamate is found at high levels in the insula and posterior cingulate, which control pain and emotion processing in the cerebrum of patients with FM [161-163].
NGF is a neuropeptide involved in the growth, maintenance, and survival of neurons and also plays an important role in pain regulation. Compared to normal controls, a significant increase in the concentration of NGF was observed in the CSF of patients with FM [163]. In a study comparing plasma BDNF and NGF levels between FM patients and healthy controls, no significant difference was observed between the two groups [164]. Further studies are needed to explore these conflicting results.
Serotonin, which is also known as 5-HT, is a monoamine neurotransmitter that plays an important role in pain control as well as mood, cognition and memory. In patients with FM, lower levels were observed than in normal controls, and were associated with pain, fatigue, and depression. This decrease in serotonin may also contribute to the development of FM [165].
Dopamine, another important neurotransmitter, has been found to be involved not only in pleasure, motivation and motor control, but also in the descending inhibitory modulation of pain in the brain [166]. Because dopamine is involved in both pain regulation and affective processing, impaired dopaminergic neurotransmission may play a particularly important role in the pathogenesis of FM.
The CSF concentration of the opioid peptide nociceptin was found to be significantly higher in FM patients than in healthy controls. Nevertheless, it was reported that FM patients had a significantly lower pressure threshold and more tender points than the control group. This may be helpful in explaining the mechanism by which the endogenous opioid system is already fully activated in FM patients, and thus exogenous opioids are ineffective [167,168].
Decreased levels of inhibitory neurotransmitters, such as serotonin, norepinephrine, and dopamine, and elevations in excitatory neurotransmitters such as SP, glutamate, and NGF are key pain amplification mechanisms. An imbalance in these neurotransmitters may be a cause of the onset of FM (Fig. 2) [163,169,170].
4) Other factors
(1) Peripheral pain generators
Local abnormalities such as osteoarthritis, enthesopathies, meniscal injury, bursitis, neuropathy, myofascial trigger points, ligamentous trigger points, or osteoarthritis of the joints and spine are common in patients with chronic widespread pain, including FM. These abnormalities act as a peripheral pain generator and can be involved in the initiation and persistence of chronic pain [171-173]. However, the role of peripheral pain generators in the onset of FM and induction of central sensitization remains a matter of debate [174].
(2) Small fiber neuropathy
Several studies have reported that FM is associated with small fiber neuropathy (SFN), which is defined as abnormally reduced epidermal nerve fiber density. FM patients without depression were significantly more likely to have small fiber dysfunction accompanied by increased cold and warm detection thresholds when compared to healthy controls [175]. In another study, a significantly higher incidence of small fiber polyneuropathy was observed in patients with FM than in healthy controls [176]. A meta-analysis of 222 FM patients reported that the prevalence of SFN in FM was 30% to 76%, and that 49% of FM patients had structural abnormalities of small nerve fibers [177].
Although SFN does not fully explain the chronic widespread pain, chronic fatigue, or mood and sleep disturbances commonly seen in FM, it can be assumed that these abnormal neuronal changes at the peripheral level may contribute to the activation of pain pathways.
(3) Muscle abnormalities
Although evidence for structural muscle abnormalities in FM is lacking, some studies have reported changes in muscle metabolism. In a study that evaluated muscle metabolism using magnetic resonance spectroscopy, patients with FM had significantly higher muscle fat content in the quadriceps muscle than the control group, while the concentrations of adenosine triphosphate (ATP) and phosphocreatinine (PCr) were significantly lower. Decreased muscle concentrations of ATP and PCr were also associated with reduced physical capacity in the leg and the hand [178]. In other studies, light microscopy and histochemical and immunoenzymatic methods did not find definitive evidence of muscle disease in the skeletal muscle of patients with FM. However, microscopic abnormalities such as empty basement membrane sleeves, many lipofuschin bodies, and other degenerative changes were observed on ultrastructural evaluations [179].
Pain Mechanisms in FM
1) Peripheral sensitization in FM
Extrinsic and peripherally generated stimuli activate nociceptors in nerve endings, and the action potentials generated reach the dorsal horn of the spinal cord through Aδ-fibers or C-fibers and are transmitted to the brain through the spinothalamic tract. When stimulation is continuous and repeated, the production of mediators (e.g., neuropeptides, proinflammatory cytokines, eicosanoids) is promoted in non-neural cells (such as mast cells, basophils, platelets, macrophages, neutrophils, endothelial cells, keratinocytes, and fibroblasts) through tissue damage or inflammatory response. Increased concentrations of these mediators form an “inflammatory soup,” which results in greater activation of nociceptors. Most of the impulses generated in the nociceptor proceed toward the spinal cord, but some are transmitted to other terminal branches and secrete several neuropeptides, including SP, at the nerve endings. The secreted neuropeptides trigger the secretion of histamine and serotonin through vasodilation, stimulation of nearby mast cells and platelets, and form a vicious cycle in which nociceptors are stimulated again through the formation of an additional inflammatory soup. This series of processes is called peripheral sensitization [143].
In patients with FM, a significant increase in mast cells in the papillary dermis was confirmed by skin biopsy (5 to 14 times more than controls). Since symptoms such as fatigue, headache, flushing, abdominal discomfort, hypotension, and tachycardia, which are common in FM, are also frequently observed in mast cell degranulation, it is assumed that there is a correlation between them. Increased mast cell degranulation in patients with FM may play an important role in peripheral sensitization [180,181]. Meanwhile, the aforementioned peripheral pain generators can further contribute to peripheral sensitization by providing additional input to the sensitized nociceptive pathway. This can be explained by some studies showing that treatment with peripheral pain generators in patients with conditions such as myofascial pain syndrome increased the pain threshold and reduced remote secondary heat hyperalgesia [182,183]. To sustain and extend central sensitization, persistent peripheral nociceptive input may be important (Fig. 2) [184].
Active management of these peripheral pain generators can help manage FM symptoms by decreasing central sensitization. However, since the symptoms of FM often do not resolve even if these peripheral factors are removed, central sensitization is assumed to play a more important role in the pathogenesis of FM.
2) Central sensitization in FM
Central sensitization is defined as the amplification of nerve signals in the central nervous system, resulting in hypersensitivity to pain [185]. Several studies have demonstrated that central sensitization by abnormal pain processing (i.e., abnormal ascending and descending pain pathways), is a key component of the pathophysiology of FM (Fig. 2). Various mechanisms are involved in this process. As a result, hyperalgesia, allodynia, and hypersensitivity to various external stimuli such as sound or light appear in FM [186-190].
(1) Abnormally activated ascending excitatory pathways in FM
Excitatory neurotransmitters such as glutamate, SP, and NGF are present at increased levels in the CSF of patients with FM [154,163,191]. In animal models of FM, superficial dorsal horn neurons are strongly sensitized, and an increase in excitatory postsynaptic input and a decrease in inhibitory input are also observed in the superficial dorsal horn [192]. This helps explain the ascending nociceptive hypersensitivity at the spine level in FM.
(2) Structural and functional changes of the brain in FM
fMRI enables the evaluation of active areas of the brain, the degree of connection between brain areas, and morphological evaluation of the brain. This is very helpful in studying the pathogenesis of FM. In a study using fMRI, when the same amount of pressure was applied, FM patients showed greater neural activation in the pain processing area of the brain than the control group [193]. The brain regions that consistently showed greater activation were the secondary somatosensory cortex, insula, and anterior cingulate cortex [194]. This suggests that patients with FM are much more sensitive to pain than healthy subjects, as the activation of pain-related brain regions is induced even with less pressure.
In patients with FM accompanied by depression, increased blood flow can be observed in the amygdala and anterior insula, which are important areas for the emotional pain response [124]. FM patients also exhibit less activation in the ventral segmental area, which controls sensory, affective, cognitive, and pain-modulatory processes, than controls [195]. This may also help explain the central sensitization of pain. In a study that evaluated brain activity caused by spontaneous chronic pain during rest, patients with FM had increased insular connectivity to the default mode network, and these changes were correlated with pain intensity [196].
In voxel-based morphometric analysis of fMRI, FM patients showed a significant decrease in total gray matter volume, and a 3.3-fold greater age-associated decrease in gray matter compared to healthy controls. Gray matter loss was more severe as disease duration increased, suggesting that FM can accelerate brain aging [197]. This gray matter volume loss was observed not only in areas related to stress and pain processing, but also in areas related to cognitive function [198]. In a study comparing FM patients and pain-free controls using proton magnetic resonance spectroscopy, FM patients showed a higher concentration of glutamate in the posterior insula than the control group [160]. In a study investigating white matter changes with diffusion-weighted imaging, patients with FM had significantly increased damage to white matter microstructure in the body of the corpus callosum associated with pain intensity than healthy controls [199]. Due to these changes observed in the brain, central sensitization is presumed to play an important role in the pathogenesis of FM.
(3) Abnormal descending inhibitory pain pathways in FM
Pain perceived by the brain is appropriately regulated through the descending inhibitory pathway. Representative descending inhibitory pathways include the serotonergic (5-HT-containing), noradrenergic, and dopaminergic inhibitory pathways. Serotonin and norepinephrine, key neurotransmitters in the descending inhibitory pathway, were decreased in the CSF of patients with FM [169,200]. A study used positron emission tomography to evaluate endogenous release of dopamine for experimental pain in FM patients and healthy controls. In the control group, dopamine was secreted from the basal ganglia during pain stimulation, but this finding was not observed in FM patients [32]. This suggests that pain suppression mechanisms are also impaired in FM.
fMRI is also useful for assessing the degree of connectivity between brain regions. In one study, the rostral anterior cingulate cortex, a brain region connected to the descending inhibitory pathway in patients with FM, exhibited reduced activity in response to pain induction [201]. In another study, patients with FM showed more abnormal resting state functional connectivity of the periaqueductal gray than healthy controls [202], and exhibited less connectivity in the brain's pain inhibitory network for pressure pain than healthy controls [203]. These findings suggest that there is an impairment of the descending inhibitory pathway for pain in patients with FM. fMRI also revealed significant impairment of brainstem/spinal cord network connectivity in patients with FM compared to healthy controls [204]. These connectivity problems make the transmission of endogenous pain inhibitory signals impossible even when the brain recognizes pain, which can aggravate central sensitization.
In another study using proton magnetic resonance spectroscopy, a decrease in the concentration of GABA, a pain inhibitory neurotransmitter, was observed in the insula of FM patients compared to healthy controls [205]. In a study using μ-opioid receptor positron emission tomography, a decrease in μ-opioid receptor binding potential was observed in regions involved in pain modulation, such as the nucleus accumbens, the amygdala, and the dorsal cingulate, in FM patients compared to healthy controls [168]. However, FM patients showed higher opioid levels in CSF than healthy controls [167]. Patients with FM generally show less response to exogenous opiates, which is presumed to be due to altered endogenous opioid receptor activity.
In an fMRI study of spinal cord and brainstem activation, patients with FM showed abnormal temporal-summation-of-second pain for repetitive heat stimuli compared to the control group [206]. This is presumed to be due to pain modulation dysfunction in FM.
Taken together, the results of several studies suggest that central sensitization plays a very important role in the pathogenesis of FM. However, it is unclear whether the ascending excitatory pathways or descending inhibitory pathways play a more important role (Fig. 2).
Conclusion
Here, we reviewed several findings related to the etiopathogenesis of FM. Our understanding of the pathogenesis of FM has improved over several decades through genetic, psychophysiological, and neuroimaging studies. FM is presumed to be caused by complex interactions of various factors, including genetic and environmental factors, neuroendocrine system abnormalities, sleep disorders, psychiatric problems, immunological abnormalities, neuroinflammation, peripheral pain generators, and SFN. Through mutual interactions, these factors can be involved in the exacerbation of various clinical symptoms, as well as the development of FM (Fig. 1).
Central sensitization in FM has been reported, with evidence suggesting enhancement of ascending excitatory pathways, suppression of descending inhibitory pathways, and structural/functional abnormalities of the brain. This leads to a decrease in the pain threshold, and FM patients suffer from chronic hyperalgesia and allodynia. It also seems to be responsible for the non-pain features of FM (Fig. 2).
Because various factors are involved in the etiopathogenesis of FM, treatments are often insufficient. It may be helpful to subclassify patients with FM according to etiopathogenesis and take a tailored approach. To achieve this, further scientific and clinical studies on the etiopathogenesis, diagnosis, and treatment of FM are needed.
Notes
Conflict of Interest
No potential conflict of interest relevant to this article was reported.