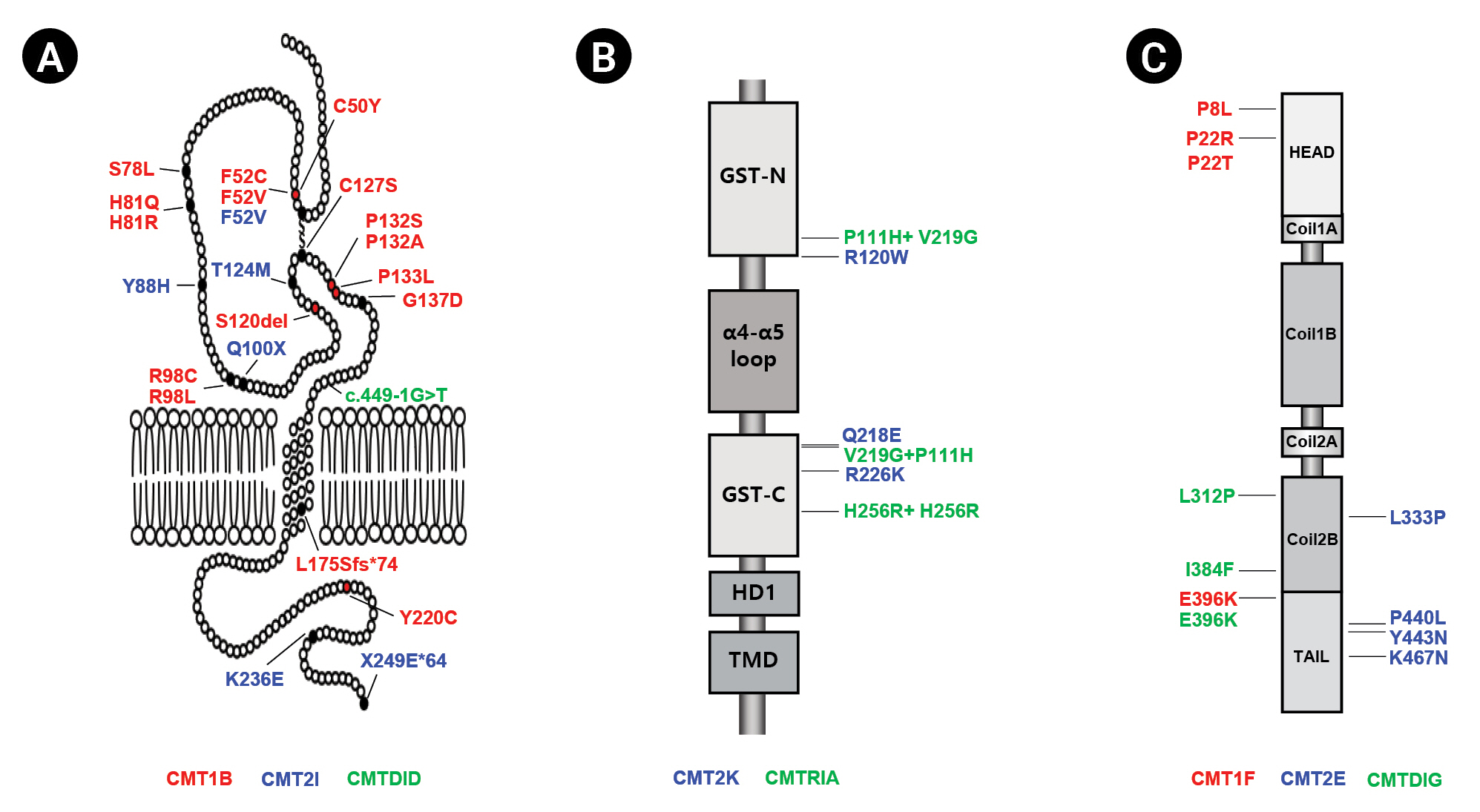
Introduction
Charcot-Marie-Tooth disease (CMT) is an inherited peripheral neuropathy that is genetically highly heterogeneous, with more than 140 different genes involved [1-3]. Classically, CMT can be divided according to clinical, histopathological, and electrophysiological findings into three types: the demyelinating type (CMT1), with a median motor nerve conduction velocity (MMNCV) below 38 m/s; axonal neuropathy (CMT2), with an MMNCV above 38 m/s; and the intermediate type (CMTDID), with an MMNCV lying between 25 and 45 m/s and nerve pathology showing axonal and/or demyelinating features [4]. Mutations in the myelin protein zero (MPZ), ganglioside-induced differentiation related protein 1 (GDAP1) and neurofilament light-chain polypeptide gene (NEFL) genes have been reported to cause all three CMT types (demyelinating, axonal, and intermediate) (Fig. 1).
Strictly expressed in myelinated Schwann cells, MPZ is a transmembrane protein that is a major component of peripheral myelin [5]. Mutations in MPZ have been reported to cause demyelinating CMT1B (MIM 118200), axonal CMT2I (MIM 607677), and intermediate CMTDID (MIM 607791) (Table 1) [6-11]. CMT1B patients generally exhibit an early onset, while CMT2I patients are characterized by a late onset [12,13]. MPZ-related CMT patients display a spectrum of diverse phenotypes, with phenotypic variations even in the same mutation. [8,14,15].
GDAP1 is mainly expressed in neurons, is located in the outer membrane of the mitochondria, and belongs to the glutathione S-transferase family [16]. Mutations in the GDAP1 gene were reported for the first time in 2002 to cause autosomal recessive (AR) CMT4A (MIM 214400) in Tunisian families [17]. Since then, GDAP1 mutations have been reported to cause axonal forms (CMT2K; MIM 607831) [18,19], axonal forms with vocal cord paresis (MIM 607706) [18] and intermediate forms (CMTRIA; MIM 608340) of disease (Table 1) [20]. GDAP1-related patients harboring AR inheritance exhibit severe clinical features with early onset, but autosomal dominant GDAP1 mutations show mild clinical symptoms with an adult onset [17,18].
NEFL is the most abundant of the three neurofilament proteins, which are major components of the axoskeleton that provide structural support for axons and regulate axon diameter. Patients with NEFL mutations exhibit a diverse phenotypic spectrum [21]. A mutation in NEFL was first reported to cause CMT2E (MIM 607684) in 2000, and several mutations were subsequently revealed to be associated with CMT1F (MIM 607734), CMTDIG (MIM 617882) (Table 1) [21-25].
Most CMT-related genes cause one CMT neuropathy subtypeŌĆödemyelinating, axonal, or intermediate neuropathy. Thus, it is noteworthy when a single gene causes multiple subtypes of CMT. In this review, we introduce the various types of CMT in a Korean cohort, caused by mutations in MPZ, GDAP1, and NEFL, and describe their clinical, electrophysiological, and genetic characteristics.
Myelin Protein Zero (MPZ)
1) Clinical diversity of MPZ-related patients
The frequency of Korean CMT families with the MPZ mutation was found to be 3.2% in all independent patients and 4.7% in CMT families without PMP22 duplicates (Table 2) [21,23,26-43]. These mutation frequencies were similar to those reported in China (3.3% and 6.4%, respectively) and Britain (3.1% and 5.1%, respectively) but lower than those reported for most other investigated ethnic groups [26,27].
The mean age at onset was 9.3 ┬▒ 10.7 years for CMT1B patients, 21.2 ┬▒ 13.7 years for CMTDID patients, and 38.7 ┬▒ 13.6 years for CMT2I patients (Table 3). The age at onset was significantly different between CMT1B and CMTDID patients (p = 0.025) or CMT2I patients (p < 0.001). However, the age at onset was not significantly different between CMTDID and CMT2I patients. Functional disability was significantly more severe in CMT1B patients than in CMT2I patients (CMT neuropathy score version 2 [CMTNS], p = 0.004, and functional disability scale [FDS], p = 0.022). The CMTNS and FDS values of the CMT1B patients were higher than those of the CMTDID patients, but the difference was not significant. When comparing the degree of disability based on CMTNS, most patients had moderate disease (53%), followed by those with severe disease (31%) and mild disease (17%) in CMT1B families. In CMTDID and CMT2I families, most patients had mild disease (75% and 80%, respectively).
2) Electrophysiological findings in MPZ-related patients
The mean MNCV of CMT1B patients was 12.2 ┬▒ 11.0 m/s, which was significantly lower than that of CMT2I patients (46.0 ┬▒ 6.6 m/s, p < 0.001) and CMTDID patients (41.3 ┬▒ 3.1 m/s, p < 0.001) (Table 3). The mean sensory nerve conduction velocity (SNCV) (7.4 ┬▒ 11.7 m/s) of CMT1B patients was significantly lower than that of CMTDID patients (18.0 ┬▒ 25.5 m/s, p = 0.021) and CMT2I patients (34.4 ┬▒ 4.3 m/s, p < 0.001). In addition, the peroneal MNCV and sural SNCV were significantly reduced in CMT1B patients compared to CMTDID or CMT2I patients. The median motor nerve compound muscle action potential (CMAP) amplitudes (6.0┬▒5.6 mV) in the CMT1B patients were significantly lower in CMTDID (12.8┬▒1.9 mV, p = 0.033) and CMT2I patients (13.0┬▒4.3 mV, p = 0.011). The peroneal nerve CMAP and median and sural sensory nerve action potential (SNAP) amplitudes were also significantly lower in CMT1B patients than in CMTDID and CMT2I patients.
Ganglioside-Induced Differentiation Related Protein 1 (GDAP1)
1) Clinical diversity of GDAP1 mutations
The GDAP1 mutation frequency rate was found to be 0.7% in all patients and 1.0% in patients negative for PMP22 duplication (Table 2). Similar frequencies have been reported in most Asian and Western countries, including Japan, China, Germany, the United States, and the United Kingdom [27-31]. However, higher frequencies of GDAP1 mutations have been reported in certain regions in Italy and Spain [32,33].
The functional disabilities and clinical characteristics were different between CMT2K and CMTRIA patients. CMT2K patients exhibited mild to moderate disabilities, with a late age at onset (19.7 ┬▒ 7.7 years), but CMTRIA patients showed severe disabilities with an early age at onset (1.7 ┬▒ 0.6 years) (Table 4). Functional disability was significantly more severe in CMTRIA patients than in CMT2K patients. The mean value of the FDS [44] was 2.3 ┬▒ 1.4 in CMT2K patients and 5.3 ┬▒ 1.2 in CMTRIA patients (p = 0.010). The mean CMTNS score [45] was 11.1┬▒6.6 in CMT2K patients and 24.7 ┬▒ 3.2 in CMTRIA patients (p = 0.011). High values of the FDS (scores of 6-7) were observed only in CMTRIA patients. In contrast, low values of the FDS (scores < 5) were observed in CMT2K patients. All three CMTRIA patients were classified in the severe category (CMTNS Ōēź 21). Foot deformities were frequent, and four patients had scoliosis. However, no wheelchair dependence, diaphragmatic weakness, vocal cord paresis, or hoarseness was observed.
2) Electrophysiological findings in GDAP1-related patients
Electrophysiological findings verified that CMTRIA patients were more severely affected than CMT2K patients. In CMT2K patients, the conduction velocity mostly did not decrease, excluding nerves with a very low amplitude. In CMTRIA patients, the decreases in CMAP and SNAP amplitudes were even more pronounced, and these parameters were not measured when nerves were explored. These results were worse in the lower extremities than in the upper extremities.
Neurofilament Light-Chain Polypeptide (NEFL)
1) Clinical diversity of NEFL mutations
The frequency of NEFL mutations was reported to range from 0.9% to 2.3% in Japanese and Chinese cohorts and in Korea (Table 2). Data on the frequency of NEFL mutations are extremely limited, though the proportion of the NEFL mutation in CMT has rarely been reported to be below 1%. Therefore, the frequency of NEFL mutations observed in East Asian countries is higher than that reported in other countries.
The prevalence of NEFL mutations was 0.44% in CMT1F patients (5/1,137), 0.35% in CMT2E patients (4/1,137), and 0.70% in CMTDIG patients (8/1,137). The age of onset was thus significantly earlier in CMT1F patients (10.2 ┬▒ 7.3 years, p = 0.013) and CMTDIG patients (12.7 ┬▒ 7.9, p = 0.007) than in CMT2E patients (24.2 ┬▒ 9.4 years) (Table 5). However, the CMTNS and FDS, as measures of disease-related disability, showed no differences among the NEFL-related CMT subtypes. Gait ataxia was identified as the most frequent symptom of NEFL-related CMT patients (78% of CMT1F patients, 50% of CMT2E patients, and 79% of CMTDIG patients). Patients were genetically tested for spinocerebellar ataxia, but no associated mutations were found. In CMT1F and CMTDIG patients, early-onset dementia was observed. Interestingly, ptosis was predominantly observed in CMT2E patients (50%).
2) Electrophysiological findings in NEFL-related patients
In CMT1F patients, the amplitudes of evoked peroneal motor responses were often markedly decreased, and the amplitudes were predominantly unrecordable in 6 of 8 patients (75%) (Table 5). However, peroneal CMAP amplitudes in CMT2E patients could not be recorded in only 1 of 4 patients (25%). Interestingly, peroneal CMAP amplitudes in CMTDIG patients (36%) occupied an intermediate position between the CMT1F and CMT2E patients. The mean MNCV of the median nerve was 16.1 ┬▒ 10.5 m/s in CMT1F patients and 47.7 ┬▒ 8.1 m/s in CMT2E patients. The mean MNCV of the median nerve was 39.6 ┬▒ 4.4 m/s in CMTDIG patients . No SNAP amplitudes of the median nerve were observed in 63% of CMT1F patients, 32% of CMTDIG patients, and 25% of CMT2E patients. Furthermore, sural SNAP amplitudes were not evoked in any of the CMT1F patients, nor were they observed in 36% of CMTDIG patients and 25% of CMT2E patients.
Conclusion
In this review, we described the clinical, electrophysiological, and genetic characteristics of various types of CMT caused by mutations in MPZ, GDAP1, and NEFL. CMT is a peripheral neuropathy with extreme clinical and genetic heterogeneity. Of the 140 causative genes for CMT and other related diseases, MPZ, GDAP1, and NEFL are the only genes that cause all three subtypes of CMT (demyelinating, axonal, and intermediate). CMT is presently incurable; however, the ongoing attempts to treat it with various drugs, dietary supplements, and increase the importance of an exact genetic diagnosis for precision medicine. Therefore, it is important to compare the genetic and clinical features of patients with MPZ, GDAP1, and NEFL mutations. Taken together, a comparison of the causative mutations and clinical features of patients with MPZ-, GDAP1-, and NEFL-related CMT will be the first step in understanding how the different types of CMT are caused, and will enable molecular genetic diagnosis.