Introduction
Neuroacanthocytosis syndromes are a group of diseases characterized by progressive basal ganglia degeneration and acanthocytosis [1]. Chorea-acanthocytosis (ChAc), a core neuroacanthocytosis syndrome, is characterized by chorea, psychiatric symptoms, and cognitive decline. It is also associated with neuromuscular abnormalities, including myopathy and axonal neuropathy. Approximately 85% of all patients with ChAc have elevated serum creatine kinase (CK) levels, termed as hyperCKemia [2], which is a nonspecific marker of muscle damage. Differentiation from other hyperCKemia-causing lesions, especially those of primary muscular diseases, is required when a patient presents with other clinical features indicative of ChAc.
Here, we describe a case of ChAc that was misdiagnosed and inappropriately treated as inflammatory myopathy (IM) due to lower extremity weakness and hyperCKemia. Furthermore, we discuss the points of differentiation between ChAc and potentially treatable acquired IM.
Case Report
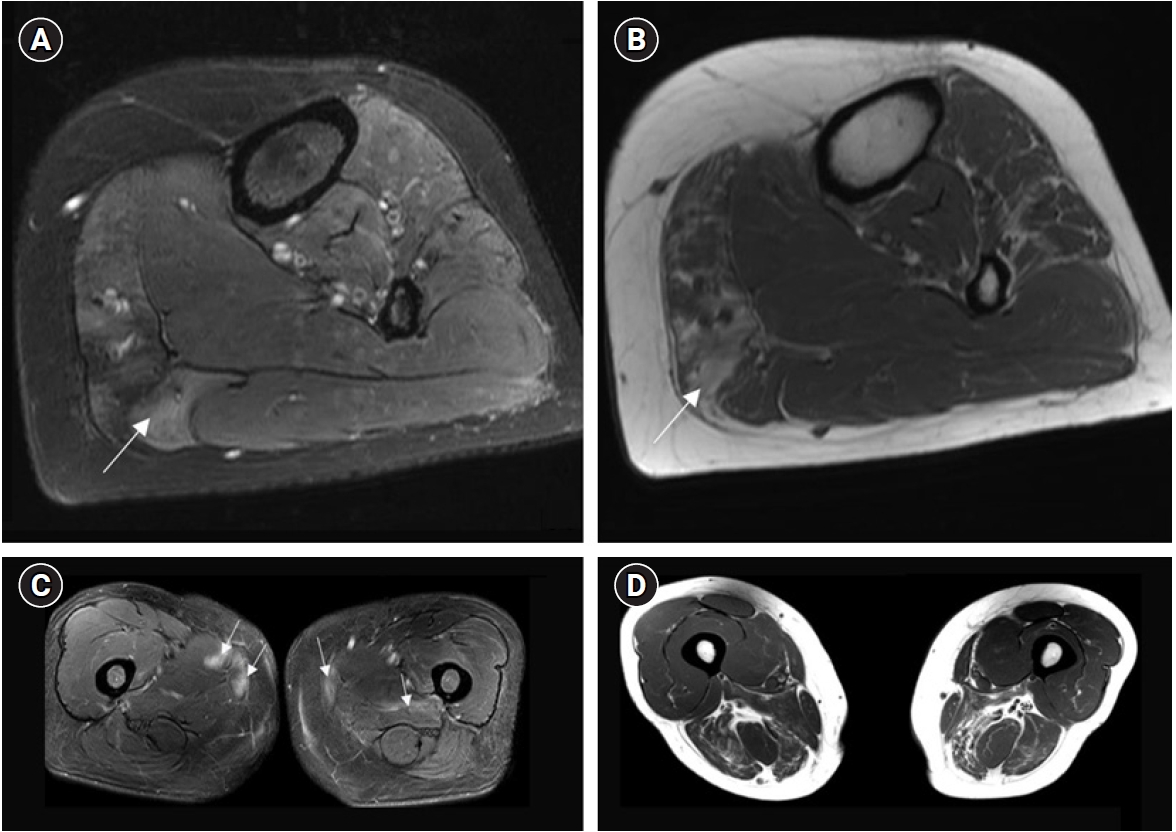
A 45-year-old female presented at a local community hospital with a 1-year history of bilateral lower extremity weakness, dysphagia, and mastication difficulty. Laboratory and electrodiagnostic testing revealed hyperCKemia and a mixed myopathic neuropathic pattern with polyneuropathy, respectively. Because of suspected myopathy, muscle biopsy was performed at the left medial head of the gastrocnemius muscle, in which magnetic resonance imaging (MRI) showed a T2 hyperintensity on the lower leg (Fig. 1A, B). She was diagnosed with autoimmune IM and treated with oral steroids and additional intravenous immunoglobulin G for 3 years. She was referred to the locomotor medicine clinic of Samsung Medical Center after showing no symptom improvement. Written informed consents were obtained.
According to her medical history, her initial symptoms were swallowing/chewing difficulty and truncal instability during walking, which appeared at the age of 43 years. She also reported tingling in both knees. She experienced difficulties in holding small objects because of the impaired fine hand movements. Her elder brother had generalized chorea and died in his early 40s.
Neurological examination revealed bilateral hip, ankle, wrist, and finger weakness without overt atrophy. She had a bilateral positive Trendelenburg sign and an uncompensated gluteus medius gait pattern. No Gower sign was observed. The anterior neck flexor and knee extensors on both sides had normal muscle strength. She had touch hypoesthesia with a stocking-glove distribution. Deep tendon reflexes were absent in all four extremities. Tongue dyskinesia and facial oromandibular dystonia were observed, and frequent tongue biting resulted in poor ingestion of solids.
Her laboratory findings included hyperCKemia (CK level, 816 IU/L; reference range, 0-170 IU/L), with acute phase reactants, antibodies for necrotizing autoimmune myositis, and HLA B51 all within normal limits. Electrodiagnostic tests revealed symmetrical sensorimotor axonal polyneuropathy. However, there was no history of systemic diseases, such as diabetes, that could cause polyneuropathy. Reduced motor unit action potential (MUAP) recruitment was observed in the muscles of the upper and lower limbs and the tongue. However, there was no denervation potential or myopathic MUAP.
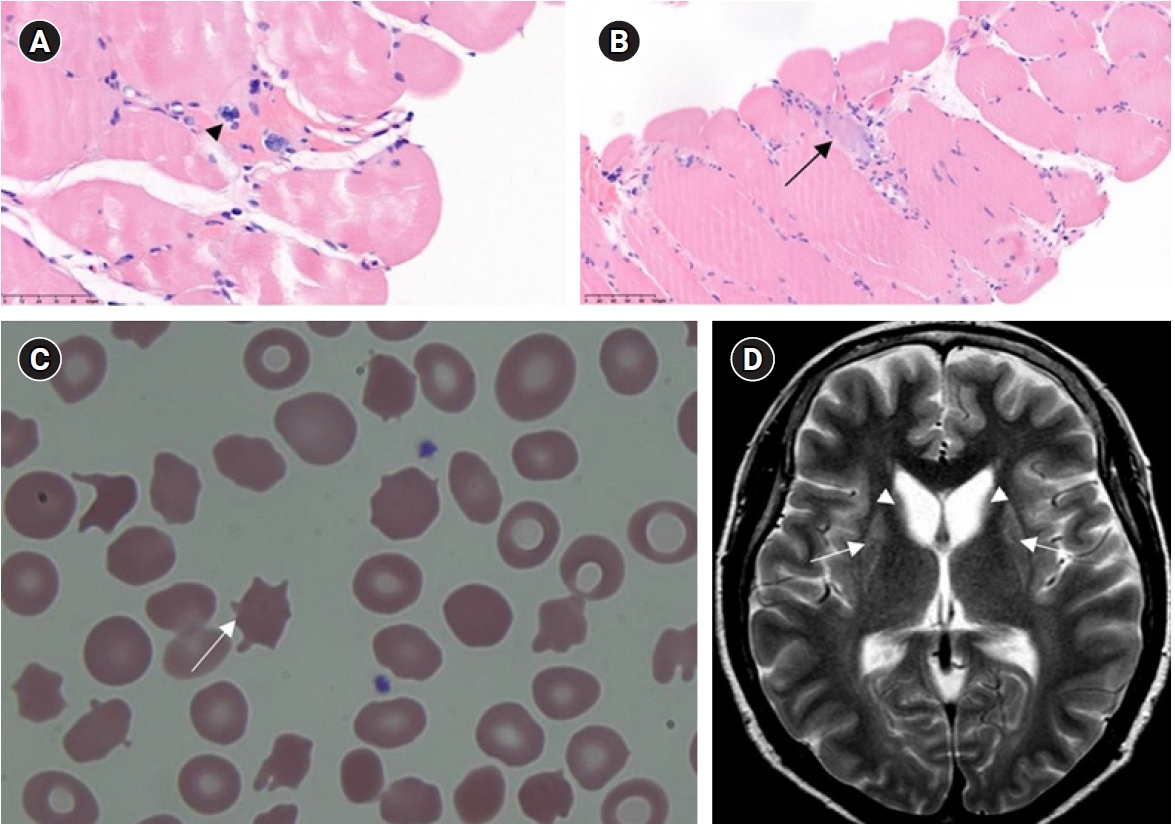
The paraspinal muscles were normal on lumbar spine MRI. Additionally, thigh MRI revealed fatty degeneration and atrophy of the bilateral hamstring and adductor magnus muscles (Fig. 1C, D). Therefore, an electrodiagnostic test was additionally performed on the muscles and patchy T2 hyperintensities were noted. Denervation potential was observed in the right gracilis and left semitendinosus muscles, with no typical neuropathic or myopathic MUAP. A reexamination of the previous muscle biopsy showed no evidence of an IM (Fig. 2A).
Considering the family history of chorea and physical and laboratory findings, neuroacanthocytosis was suspected. Peripheral blood smear and brain MRI were performed, which revealed acanthocytosis and bilateral caudate nucleus atrophy with T2 hyperintensity in the bilateral basal ganglia (Fig. 2B, C).
Next-generation sequencing for dystonia revealed two likely pathogenic variants of the vacuolar protein sorting 13 homolog A (VPS13A) gene. The patient was finally diagnosed with ChAc [1]. She was administered haloperidol, following which her feeding dystonia and lingual dysarthria improved slightly, but with no change in hyperCKemia and gait disturbance.
Discussion
ChAc is an autosomal recessive neurodegenerative disorder. Most patients with ChAc present with choreatic movement disorders, including orofaciolingual dystonia, and various neuromuscular manifestations, including muscle weakness, atrophy, and areflexia [1]. Because its neuromuscular manifestations have not been sufficiently investigated, it can be initially mistaken as a primary myopathy. In early-stage ChAc, abnormal hyperkinesia is not severe. Proximal and distal muscle weakness usually appear late (age, 20-40 years), often with accompanying hyperCKemia. These characteristics may lead to erroneous diagnoses, particularly autoimmune IM.
Based on the characteristics of this case, several differentiating points between ChAc and IM may be suggested. First, the distribution of muscle weakness is different; the latter generally shows axial and symmetric proximal girdle muscle weakness. Although this patient had bilateral hip abductor muscle weakness, the anterior neck flexor and knee extensor had normal strength, and the Gower sign, which is a hallmark of proximal girdle muscle weakness, was not found. Moreover, lumbar spine MRI did not show fatty atrophy of the lumbar paraspinal muscles, which is often seen in muscular dystrophies. Second, sensory hypoesthesia with a stocking-glove distribution was observed, which was later confirmed as symmetric axonal polyneuropathy through a nerve conduction study. Third, no definite evidence of myopathy was found on the electrodiagnostic test. In a study by Rampoldi et al. [2], 67% of patients with ChAc showed neuropathic features in an electrophysiological study, but none showed myopathic features. This could differentiate ChAc from IM, in which the electromyographic characteristics of myopathy, such as short duration, small amplitude, polyphasic MUAPs, and an early complete recruitment pattern on volition are generally observed. Fourth, MRI showed asymmetrical focal patchy involvement of several thigh muscles. In contrast, the muscle involvement in IM is usually diffuse and symmetric [3]. Finally, chorea is frequent in neuroachantocytosis, and orofaciolingual dystonia is a specific finding of ChAc. In this case, mandibular symptoms played an important role in the differential diagnosis.
Previous studies have reported that the muscle histology of ChAc (increased number of internal nuclei, increased fiber size variation, and mild endomysial fibrosis) is consistent with that of moderate myopathy. However, these myopathic findings may be secondary to chronic denervation changes, and the main findings could be small-group atrophy, increase of small fibers on diameter analysis, and frequently angulated fibers [4]. Therefore, muscle biopsy findings of ChAc are clearly different from those of IM, with the latter showing perimysial and endomysial mononuclear cell infiltrates (polymyositis) or perivascular inflammation, perifascicular atrophy of muscle fibers, and endomysial and perimysial vasculitis (dermatomyositis). Our case showed only nonspecific myopathic changes.
The protein chorein is not expressed in ChAc, as seen in Western blot analysis, and confirmatory diagnosis is made by identifying pathological variants of the VPS13A gene that encodes chorein. The exact roles of chorein in neurodegeneration and the development of hyperCKemia are not fully understood; however, it appears to be involved in the polymerization of actin; hence, its dysfunction can lead to cell membrane destruction, leading to changes such as abnormal erythrocyte shape and muscular and nerve damage [5].
In conclusion, a typical case of neuroacanthocytosis initially misdiagnosed as an IM has been reported. Careful clinical and laboratory evaluations are warranted to rule out neuroacanthocytosis in suspected myopathies presenting with chorea and peripheral neuropathy. Although rare, it is necessary to consider ChAc as the cause of hyperCKemia [5]. Early differential diagnosis can prevent unnecessary treatment with steroids or immunomodulating agents.