Introduction
Acute intermittent porphyria (AIP) is a rare genetic disorder that induces severe disabilities. The development of symptoms is associated with various exacerbating factors, from medications to poor oral intake or stress, in patients with genetic factors. The clinical manifestations of AIP include abdominal pain, neurological symptoms, and sensorimotor polyneuropathy. Considerable recovery can be expected in cases that receive early diagnosis and treatment [1,2].
Porphyric neuropathy is typically described as a motor-dominant axonal neuropathy with preceding abdominal pain (from a few days to weeks). Its symptoms, signs, and results of cerebrospinal fluid (CSF) analysis are similar to those of Guillain-Barré Syndrome (GBS) or Critical illness polyneuropathy (CIP). Clinical features such as asymmetric, proximal dominant weakness, accompanied with psychosis or abdominal pain, as well as axonal-type polyradiculopathy or polyneuropathy verified by nerve conduction studies, can be used for differentiating porphyric neuropathy from GBS or CIP [3]. However, AIP is rare disease that is difficult to diagnose at the first inspection of clinical data; therefore, patients with AIP rarely receive a diagnosis at an early disease stage. Here, we described the clinical course in a patient with AIP that was first reckoned as other polyneuropathies.
Case report
A 23-year-old unmarried woman without any medical history presented to the hospital with cramping abdominal pain on February 11, 2018. An abdominal computed tomography (CT) scan showed two sites of intussusception. She underwent laparoscopic manual reduction and postoperative care with limited oral intake. She was discharged from the hospital 10 days following the intervention.
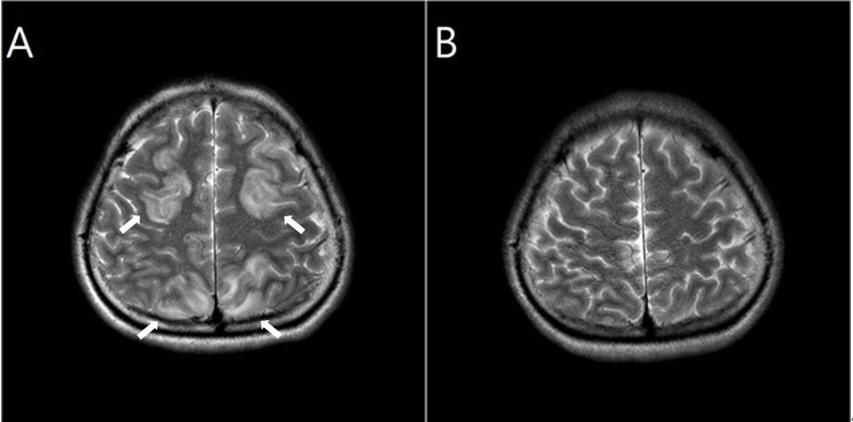
Six hours after being discharged, she experienced a sudden deterioration in her consciousness level and suffered four serial convulsions lasting for 3-5 min each. The convulsions were of a generalized tonic-clonic type. Brain CT and CSF tapping were normal, but the patient’s condition continued to deteriorate with the requirement for admission to the intensive care unit (ICU), intubation, mechanical ventilation, and tracheostomy. Magnetic resonance imaging (MRI) was performed and posterior reversible encephalopathy syndrome was observed without a definitive etiology (Fig. 1). Tests using an anti-nuclear antibody; anti-neutrophil cytoplasmic antibodies; and anti-GM1, GD1b, and GQ1b antibodies were performed, but no positive signals were detected.
The onset of motor weakness in both the upper and lower limbs was noted on the 23rd postoperative day (POD). Physical examination revealed that her motor power was of Medical Research Council (MRC) grade 2/5, with progressive deterioration to grade 1/5 1 day later; this was accompanied with the absence of all deep tendon reflexes in all limbs. The sensory functions of all limbs, as assessed using both pinprick and light-touch tests, were also impaired compared with the cheek. A nerve conduction study (NCS) revealed the presence of severe sensorimotor axonal polyneuropathy, with no response in a routine NCS of the motor and sensory nerves in the median, ulnar nerves, motor NCS of deep peroneal, posterior tibial nerves, and sensory NCS of superficial peroneal and sural nerves. The patient was diagnosed with peripheral polyneuropathy such as GBS or CIP, and was treated with intravenous immunoglobulin administration for 5 days, starting on POD 31, with no definitive effect [4].
Abdominal pain, neurological symptoms, and unexplained severe polyneuropathy raised the suspicion of AIP. For confirming this diagnosis, urine porphobilinogen, delta-aminolevulinic acid, coproporphyrin, porphyrin, and uroporphyrin levels, as well as blood porphyrin concentration, were assessed. A urine color change was also observed after 24 h of exposure to sunlight (Fig. 2). Ten days following the suspicion of AIP, a diagnosis of this condition was confirmed by urine porphobilinogen levels (102.01 mg/day) on POD 40 (Table 1). Two 4 day courses of intravenous hemin were administered after the diagnosis, for proper management of the disease. None of her extremities exhibited a change in power after hemin injection.
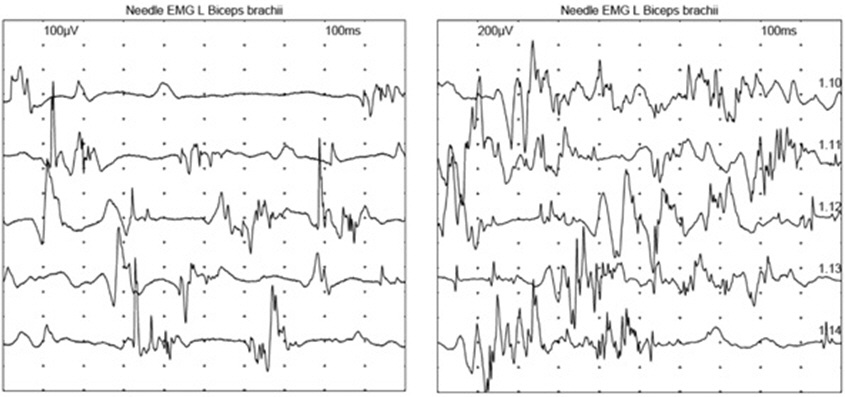
A follow-up NCS was performed 2 months after the onset of quadriplegia, without any change in muscle strength or sensory functions. There were no responses in any of the sensory and motor nerves tested. We observed abnormal spontaneous activities in all muscles tested, and no motor unit action potential. Three months later, the motor power of the proximal upper limbs and lower extremities was improved, to an MRC grade of 3/5. Although there was no improvement in the NCS findings, some regeneration signs, that is, polyphasic motor unit action potential(MUAP) and 2-3 different MUAPs were recruited, were observed in the left biceps brachii, which were absent in the tests performed 3 months previously (Fig. 3). Other muscles tested, which were right first dorsal interossei, rectus femoris, tibialis anterior showed no regeneration signs. These muscles showed no motor unit action potentials in both studies.
Discussion
Acute intermittent porphyria is a type of acute hepatic porphyria that usually presents with gastrointestinal, neurological, and psychiatric symptoms. The common clinical manifestations of this condition include severe abdominal pain accompanied with nausea, vomiting, and ileus; and neurological symptoms, such as seizure, PRES, chronic neuropathic pain, psychosis, and motor-dominant polyneuropathy [5,6]. It is well known that antiepileptic medications, which induce hepatic cytochrome P450 enzymes, act as exacerbating factors of AIP, as do etomidate, ketamine, NSAIDs, and rifampin. Glucose acts as a suppressor of delta-aminolaevulinic acid synthase expression, which participates in the heme biosynthetic pathway. Circumstances such as starvation, which may lead to caloric deficiency, also exacerbate AIP, and can trigger an acute attack [1]. In the present case, phenytoin, valproic acid, topiramate, and etomidate were administered during ICU management, and starvation after laparoscopic operation might also have contributed to the exacerbation of her condition. These drugs and circumstances elicited recurrent acute attacks with neurological symptoms. The clinical manifestations of abdominal pain, seizure, PRES, chronic neuropathic pain, tachycardia, and LFT elevation accompanied the sensorimotor polyneuropathy in our patient. These neurological and gastrointestinal symptoms are extraordinary in general neuropathies.
Porphyric neuropathy mostly presents as a primary motor neuropathy, usually sparing sensory function. Onset distribution is important to rule out other neuropathies. The disease starts in the upper extremities in 50% of patients, whereas proximal muscles, rather than distal ones, are involved in 80% of cases [6-8]. But in this case, symmetric sensorimotor involvement was found in early stage. It suggests that we should also consider porphyric neuropathy as one of the diagnosis even in patient with symmetric sensorimotor polyneuropathy. To the best of our knowledge, no clear severity criteria have yet been established. In the present case, we assumed the presence of a severe form of neuropathy in both the upper and lower extremities; moreover, facial and bulbar symptoms were involved with sensory loss. Those symptoms occurred progressively, one by one; thus, the clinical features might be different if early treatment is administered.
Nerve conduction studies and electromyography show no pathognomonic findings for porphyric neuropathy; however, these tests remain important as differential diagnostic tools. In general, porphyric neuropathy shows axonal-type neuropathy in nerve conduction studies, while sensory conduction is often spared. This can be an important clue to differentiate this disease from GBS, which generally presents with a demyelinating type, rather than an axonal one, even though it can also be of axonal type. Some limitations are still existing, such that acute motor axonal neuropathy (AMAN) type is prevalent among Far East Asian, and that severe cases without any action potentials are difficult to specify [9]. Unlike most of other cases, a follow up study was conducted after 3 months, and we could find out some regeneration signs as symptoms improved [3,6-8]. With more studies, it could provide some chances to follow up NCS, EMG as a prognostic tool that might be correlated with clinical features.
Regarding perspective of prognosis, neuropathy improved in a slower manner compared with other symptoms. Moreover, it is related to the magnitude of axonal degeneration. The prevention of recurrent attacks is also an important issue. Weakness and sensory impairment improve over several months, and sometimes incomplete recovery of motor function is observed. Pischik et al. mentioned the severity of abdominal pain, muscle weakness degree, duration of mechanical ventilation, bulbar palsy, consciousness impairment, and hyponatremia as prognostic factors in neuropathy [10]. In our case, mot of factors were severely involved, which helped predict a poor prognosis for this patient. The patient could gait a few steps alone and was able to roller-walker gait with supervision 1 year later.
Acute intermittent porphyria may cause severe peripheral axonal polyneuropathy. Porphyric neuropathy should be considered as a differential diagnosis in patients with an acutely progressing severe polyneuropathy for better prognosis.