Introduction
Hereditary neuropathy with liability to pressure palsy (HNPP) is an autosomal dominant peripheral nerve disorder caused by a 1.5 Mb deletion within chromosome 17p11.2 [1]. It is typically diagnosed in adolescence [1]. Commonly affected nerves include the median nerve at the wrist, ulnar nerve at the elbow, radial nerve in the arm, peroneal nerve at the fibular head, or other sites sensitive to pressure. In addition, other clinical phenotypes of the disorder include progressive polyneuropathy, chronic polyneuropathy, Charco-Marie-Tooth disease-like symptoms, and chronic inflammatory demyelinating polyneuropathy (CIDP)-like disorder [2].
There has been one report of an HNPP patient that presented clinically with progressive muscular atrophy (PMA) after a 40-week follow-up. However, the author mentioned that their case could involve a variant of HNPP with amyotrophic lateral sclerosis (ALS) if, upon further follow-up, upper motor neuron (UMN) dysfunction is identified [3]. Here, we present a case of HNPP that progressed like PMA throughout a 7-year follow-up period. The patient eventually died owing to bulbar dysfunction and respiratory insufficiency 7 years post-diagnosis.
Case Report
We report a case involving a 60-year-old male patient who presented with a 6-month history of painless weakness in his left upper extremity. The patient had no history of diabetes, incontinence, dysphagia, or any family history of similar illnesses. An initial physical examination revealed mildly generalized atrophy and medical research council (MRC) grade 4 motor weakness within his left upper arm without any sensory symptoms. Deep tendon reflexes were decreased at his biceps, triceps, knee, and ankle tendons, and no UMN dysfunction was observed. To identify the likely cause of symptoms, an electrodiagnostic study, C-spine, and brain magnetic resonance imaging (MRI) were performed.
Nerve conduction studies (NCS) revealed mixed sensori-motor polyneuropathy, which were mainly demyelinating. Multiple entrapment neuropathies of the median nerve at the right wrist and in the ulnar nerve at the left elbow were observed (Table 1). Needle electromyography showed denervation potentials in muscles of the left upper limb and polyphasic, or large amplitude motor unit action potentials, in both upper limbs and the left lower limb. There were no significant findings observed regarding paraspinal and facial muscle test (Table 2). The somatosensory evoked potential produced by median nerve stimulation was normal. Imaging studies revealed no remarkable findings, aside from disc protrusion at C4-5, degenerative spondylosis via cervical MRI and micro-angiopathy of periventricular white matter via brain MRI. Based on observed polyneuropathy and multiple entrapment neuropathies, further study to rule out HNPP was recommended, but the patient refused at the time.
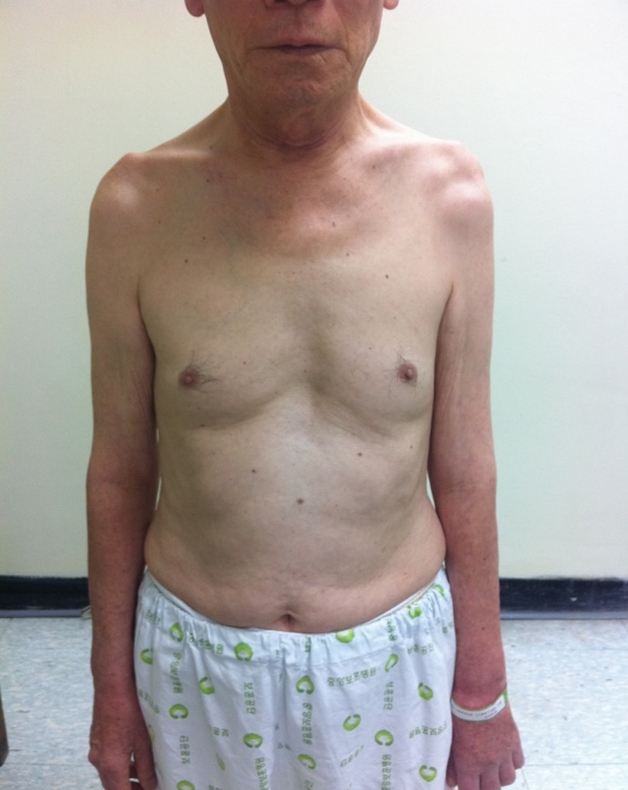
One year and eight months after his first visit, the patient experienced worsening weakness of his left arm to MRC grade 3 and newly developed weakness in his right upper limb (MRC grade 3+) 2 years post-disease onset (Fig. 1) We received the patientŌĆÖs consent form about publishing all photographic materials. Since the patient agreed to be further examined, laboratory tests, antibody testing for various autoimmune diseases, a cerebrospinal fluid (CSF) test, and genetic testing for HNPP were performed. Laboratory test results, including antibody testing for various autoimmune diseases and an anti-GM1 antibody test, were negative. Additionally, CSF results were all within normal range, with the exception of a 58.8 mg/dL CSF protein concentration, which was slightly elevated (normal range, 15-45 mg/dL). The genetic assessment revealed a heterozygous deletion of the gene encoding PMP22, which confirmed the HNPP diagnosis. After the patient received his results, an additional genetic study revealed that his youngest daughter also harbored a heterozygous deletion of the gene encoding PMP22.
Six years post-diagnosis, weakness of both upper arms continued to progress and extended to both lower extremities. The patient had no UMN dysfunction or no remarkable findings on additional C-spine and L-spine enhanced MRI study which could reveal alternative causes of the slowly progressive motor weakness. NCS revealed aggravation due to PMA (Table 3). Moreover, needle electromyography revealed newly detected fibrillation and positive sharp waves on cervical, thoracic, and lumbar paraspinal muscles (Table 4).
Seven years post-diagnosis, the patient experienced swallowing difficulty. A video-fluoroscopic swallowing study revealed decreased laryngeal elevation with moderate amount of vallecular and pyriform sinus residue. In addition, the patient required continuous positive airway pressure due to nighttime dyspnea and was not able to perform daily activities without full assistance. His muscle weakness continued to worsen to the point that he required a mechanical ventilator as a result of respiratory dysfunction. Despite these challenges, he signed a do-not-resuscitate form and died in the seventh year.
Discussion
The patient described here had an unusual HNPP disease phenotype. He presented with asymmetric left upper arm muscle weakness and atrophy and demyelinating polyneuropathy with multiple entrapment neuropathies. Genetic analysis revealing a deletion within the PMP22 gene was used to diagnose HNPP. Additionally, the absence of UMN dysfunction, reduced reflexes indicating lower motor neuron (LMN) dysfunction, unremarkable findings from image studies and fibrillation potentials, the presence of positive sharp waves, large polyphasic motor unit potential with reduced recruitment in clinically affected and non-affected areas in needle electromyography indicated possible motor neuron disease (MND) such as PMA.
Typically, HNPP presents as recurrent sensory and motor neuropathy in a single nerve with wax and wane symptoms that occur in adolescence [1]. Other studies have shown various atypical phenotypes including CIDP that resembles polyneuropathy [4], symptoms of generalized weakness and muscle cramps [5], muscular pain [6], or low back pain with radicular symptoms [7]. Few cases have revealed that HNPP may be associated with MND such as ALS or PMA [3,8]. In contrast with other reports, the first episode experienced by our patient occurred at the age of 60, and there was no history of transient muscle weakness in his adolescence. Additionally, his first episode included progressive asymmetrical left upper arm muscle weakness, rather than transient muscle weakness. Further, the muscle weakness continued to worsen over time, and slowly spread to his other limbs.
A previous report described a patient with progressive muscle weakness, dysphagia, hand tremor and distal region dysesthesia who was diagnosed with two rare diseases: spinobulbar muscular atrophy (SBMA) and HNPP [9]. Similarly, our patient also possessed slowly progressive muscle weakness and swallowing difficulty at the final stage of disease. However, our patient did not experience hand tremors, facial atrophy, or fasciculation nor did he complain of any bulbar symptoms which would have caused us to initially doubt SBMA. Further, facial and neck muscle atrophy and swallowing difficulty were identified in the seventh year post-symptom onset and likely occurred as a part of disease progression.
Myelin protein, PMP22, controls Schwann cell proliferation and apoptosis, which are needed to produce compact myelin [10]. PMP22 deletion and/or cumulative nerve entrapment could lead to secondary axonal loss and LMN apoptosis to produce PMA, since the expression of PMP22 mRNA occurs within LMNs of the spinal cord and brain stem [10]. However, only one case report has described HNPP with PMA, and it involved a relatively short, 40-week follow-up [3]. And since the follow-up period was short, the author stated that the possibility of UMN dysfunction could not be ruled out [3]. In comparison, the weakness experienced by our patient continued to slowly progress for seven years, and no UMN dysfunction was identified until his death. Swallowing difficulty and dyspnea occurred, which indicated the possibility of bulbar dysfunction on later stage. Therefore, our case, which included a long-term follow-up, revealed late onset HNPP that presented like slowly progressive PMA. The patient died because of bulbar dysfunction and respiratory insufficiency.
In conclusion, a review of our case indicates that the spectrum HNPP clinical presentation may be much broader than previously known. Our case fell into neither a well-known nor atypical spectrum of HNPP. Therefore, the description of this case may help expand our knowledge of the spectrum of HNPP. Further review studies involving a larger series of cases may be needed to fully define clinical features of HNPP.